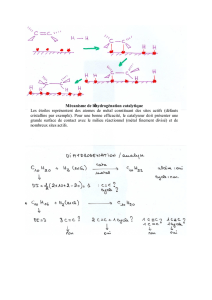
Synthèse asymétrique: Réactions et méthodes énantiosélectives

La synthèse
asymétrique

Synthèse asymétrique
Chapitre consacré au contrôle de la stéréochimie absolue, que ce soit
par synthèse « asymétrique » (énantio ou diastéréospécifique) ou par
séparation des énantiomères ou des diastéréoisomères.
En général, on désigne sous le nom de synthèse asymétrique une
addition énantiosélective, à savoir, la transformation réussie de
composés de départ achiraux en produits énantiopurs.
Note: si le composé de départ possède déja un centre chiral et que l'on
transforme un centre pro-chiral par synthèse asymétrique, on obtiendra
deux diastéréoisomères.

Définition:
face « Ré » et « Si » d'une cétone ou un aldéhyde prochiral
face « Ré » et « Si » d'un alcène
Diagramme d'énergie pour l'obtention de deux énantiomères
Diagramme d'énergie pour l'obtention de deux diastéréoisomères
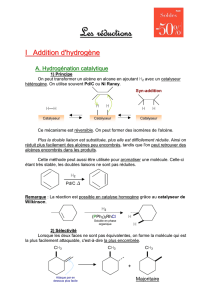
I. Introduction

Méthode Avantages Inconvénients
résolution obtention des 2 énantio. rdt max de 50%
réactif chiral svt TB ee, améliorables peu réactifs utilisables
par recristallisation svt petit nbre substrats
catalyseur chiral économique: petites peu réact. vraiment
quant. et recyclables réussies
fond commun chiral ee 100% garanti un seul enantio. obtenu
auxiliaire de chiralité svt TB ee, améliorables étape supplémentaire:
par recristallisation intro et élim. auxiliaire
« Dérivatisation » inversion de configuration avec conservation ee
Résumé des méthodes de S.A.

1) Diagramme d'énergie d'une synthèse avec obtention de 2
énantiomères
2) Diagramme d'énergie d'une synthèse avec obtention de 2
diastéréoisomères
Enantiomères et diastéréoisomères
diagrammes d'énergie
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
1
/
55
100%