Cours Chimie SP2 2016-17 [Mode de compatibilité]

Oxydoréduction et Cinétique Chimique
M 2301
Cours/TD : 5 séances de cours/TD de 2h
TP : 4 TP de 3h30
Evaluation (2*DS+1*CC+1*CR)
1 DS de 1h30
1 CC
1 note de CR de TP
Enseignants TP
Fabien Giroud et Elodie Bidal
Elodie Bidal
Bureau 501
elodie.bidal@univ-grenoble-alpes.fr
22
Sources bibliographiques
« Oxydoréduction et Cinétique Chimique S2 » – Laurent Dessemond
IUT1 de Grenoble – Département Mesures Physiques.
« Techniques d’Analyses Chimiques SP1 » – Claude Roux
IUT1 de Grenoble – Département Mesures Physiques.
« Equilibres et réactivité chimiques » – Jacques-E. Moser
Ecole Polytechnique Fédérale de Lausanne - Section de chimie et de
génie chimique.
« Chimie Physique » – Paul Arnaud – Edition Dunod
Programme du cours
Chapitre I – L’équilibre rédox
Chapitre II – Cinétique chimique
t= 2h
Chapitre I – L’équilibre rédox
I. Les transferts électroniques
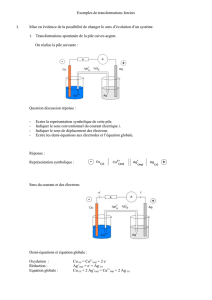
Dépôt de cuivre Cu(s) sur la lame
de zinc
- Désagrégation de la lame de zinc
- Couleur bleue moins intense
Solution de sulfate
de cuivre CuSO4
(bleue)
Lame de zinc
t= 0
I.2 Interprétation
22
2
1
2
1
2
2
réducteuroxydant
oxydantréducteur
CueCu
eZnZn
↔+
+↔
−+
−+
Réduction de Cu2+ Couple Redox : Cu2+ / Cu
Oxydation de Zn Couple Redox : Zn2+ / Zn
Equation globale d’oxydoréduction
I.1 Expérience
⇒transfert de deux électrons entre Zn et Cu2+
CuZnCuZn +↔+
++ 22

5
Une oxydation correspond à une perte d’électrons; une réduction à un gain
d’électrons.
Un oxydant capte les électrons tandis qu’un réducteur cède les électrons.
Une réaction d’oxydoréduction est une réaction d’échange (total ou partiel)
d’électrons. Elle fait intervenir 2 couples rédox.
L’équation-bilan de la réaction d’oxydoréduction est obtenue en faisant l’addition
des deux demi-équations électroniques associées aux réactions électrochimiques
dans lesquelles apparaissent les électrons.
I.3 Définitions
(1)
(2)
22
2
1
2
1
2
2
réducteuroxydant
oxydantréducteur
CueCu
eZnZn
↔+
+↔
−+
−+
6
Zn cède plus facilement les électrons que Cu :
=> Zn est un réducteur plus fort que Cu.
Cu2+ capte plus facilement les électrons que Zn2+ :
=> Cu2+ est un oxydant plus fort que Zn2+.
Entre 2 couples rédox, la réaction spontanée rédox s’effectue toujours avec l’oxydant
le plus fort et le réducteur le plus fort.
Cas des transferts électroniques partiels (liaison covalente polaire)
Exemple : H2(g) + Cl2(g) ----------> 2HCl(g) H
δ
+----Cl
δ
-
I.3 Définitions (suite)
CuZnCuZn +↔+
++ 22
Dans l’équation-bilan de la réaction d’oxydoréduction (ou rédox), les électrons
n’apparaissent plus.
(3) = (2) + (1)
7
Ecriture générale d’une réaction électrochimique :
réduction (→)
O + n e-↔ R
oxydation (←)
⇒mise en jeu du couple redox O/R.
Ecriture générale d’une réaction d’oxydoréduction :
Couple redox O1/R1:O1+ n1e-↔ R1 (1)
Couple redox O2/R2: R2 ↔O2+ n2e-(2)
⇒n2O1+ n1R2↔ n2R1+ n1O2 n2(1) + n1(2)
Remarques :
- les électrons n’apparaissent pas dans l’écriture d’une réaction d’oxydoréduction :
⇒le nombre d’électrons cédés est égal au nombre d’électrons captés.
- conservation de la matière et électroneutralité respectées quelle que soit la
réaction chimique.
I.4 Formalisme général
8
II. Le nombre d’oxydation
Le nombre d’oxydation (ou n.o.) permet de déterminer l’état d’oxydation d’un
élément soit à l’état atomique, soit inclus dans un ion ou une molécule.
C’est un nombre qui indique l’importance de la perte ou du gain d’électrons de
l’élément par rapport à l’atome neutre.
Il se note en chiffres romains.
II.1 Définition
II.2 Calcul du nombre d’oxydation
Convention
- atome isolé : n.o. = 0
- molécule d’un corps simple, par exemple H2, O2, Cl2, N2n.o.(H) = 0 ; n.o.(O) = 0 ;
- ion simple : n.o. = la charge de l’ion
- ion polyatomique : la somme* des n.o. des éléments = charge de l’ion
- molécule : la somme* des n.o. des éléments = 0
*Il faut tenir compte des indices affectant chaque atome.

9
II.2 Calcul du nombre d’oxydation
- Exemples :
Cu => n.o.(Cu) dans Cu = 0
H2=> n.o.(H) dans H2= 0
Zn2+ => n.o.(Zn) dans Zn2+ = +II
SO42- => n.o.(S) + 4*n.o.(O) = –II
H2O => 2*n.o.(H) + n.o.(O) = 0
Valeurs des n.o. des éléments les plus courants engagés dans un édifice
moléculaire ou ionique
hydrogène : n.o.(H) = +I exemple : HCl
oxygène : n.o.(O) = -II exemple : H2O
métaux alcalins : n.o. = +I exemple : Na2O
métaux alcalino-terreux : n.o. = +II exemple : CaO
Exceptions :
-dans MgH2: no(H) = - I
-dans H2O2: no(O) = - I
10
II.2 Calcul du nombre d’oxydation (suite)
Exemples d’application :
HBr : no(H) + no(Br) = 0 SO42- : no(S) + 4 x no(O) = - II
no(H) = + I no (O) = - II
⇒no(Br) = - I ⇒no(S) = + VI
Remarque :
Dans un couple redox, le nombre d’oxydation de l’oxydant est toujours supérieur au
nombre d’oxydation du réducteur.
Nombre d’oxydation moyen :
Hypothèse : combinaison chimique contenant plus d’une unité d’un élément :
Mn3O4:
3 x no(Mn) + 4 x no(O) = 0
no(O) = - II
⇒
⇒nombre moyen entre + II (Mn2+) et + III (Mn3+)
( )
3
VIII
Mnno +=
11
Oxydation : n.o.
Réduction : n.o.
Application des n.o. :
Reconnaître une réaction rédox d’une réaction acido-basique.
II.2 Calcul du nombre d’oxydation (suite)
0
22
0
CuZnCuZn
IIII
+↔+
++
++
0
22
0
2
2
1
2OFHOHF
III
+↔+
−−
Oxydation
Réduction Oxydation
Réduction
∆n.o.(F) = -1 - 0 = -1 pour 1 atome de F
∆n.o.(O) = 0 - (-2) = +2 pour 1 atome de O
2*∆n.o.(F) + ∆n.o.(O) = 0
Pas de variation du n.o. dans une réaction
acido-basique quel que soit l’élément.
II
I
I
IIIII
OHFOHFH
−+
+
−−
−+−+
+↔+
32
12
III.1 Equilibrer une demi-équation (réaction électrochimique)
a) En milieu aqueux, sans utiliser les n.o.
MnO4-/Mn2+
- écrire l’oxydant et le réducteur d’un même couple de part et d’autre de la double
flèche (oxydant à gauche, réducteur à droite):
MnO4-↔ Mn2+
- équilibrer les atomes autres que H et O ;
- équilibrer les atomes d’oxygène en rajoutant autant de molécules d’eau H2O que
nécessaire ;
MnO4-↔ Mn2+ + 4H2O
- équilibrer les atomes d’hydrogène en rajoutant autant d’ions H+que nécessaire :
MnO4-+ 8H+↔Mn2+ + 4H2O
- équilibrer les charges électriques : MnO4-+ 8H++ 5e-↔Mn2+ + 4H2O
b) En utilisant les n.o.
- identification de l’élément dont le nombre d’oxydation varie ;
- calcul des nombres d’oxydation de l’oxydant et du réducteur du couple redox ;
- écriture de la réaction électrochimique dans le sens de la réduction ;
- respect de la conservation de la matière et de l’électroneutralité.
III. Equilibrer une réaction rédox

13
III.2 Equilibrer une réaction d’oxydoréduction
- écriture de la réaction électrochimique associé à chaque couple redox ;
- égalisation des coefficients stoechiométriques des électrons pour les deux
réactions électrochimiques ;
- combinaison des réactions électrochimiques alors obtenues afin que les électrons
n’apparaissent plus.
Oxydation de Fe2+ par MnO4-en solution aqueuse acide (couples mis en jeu :
Fe3+/Fe2+et MnO4-/Mn2+) :
Fe3+(aq) + e-↔ Fe2+(aq) (1)
MnO4-(aq) + 5 e-+ 8 H+(aq) ↔ Mn2+(aq) + 4 H2O(l) (2)
⇒MnO4-(aq) + 8 H+(aq) + 5 Fe2+(aq) ↔ Mn2+(aq) + 5 Fe3+(aq) + 4 H2O(l)
Réduction de Cl2par Zn (couples mis en jeu : Cl2/Cl-et Zn2+/Zn) :
Cl2(g) + 2 e-↔ 2Cl-(aq) (3)
Zn2+(aq) + 2 e-↔ Zn(s) (4)
⇒Cl2(g) + Zn(s) ↔ Zn2+(aq) + 2 Cl-(aq)
III. Equilibrer une réaction rédox (suite)
(2) - 5x(1)
(3) - (4)
14
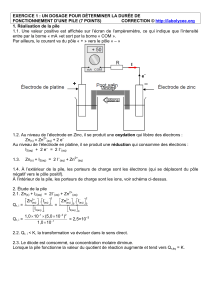
IV. Pile électrochimique
IV.1 Electrode ou demi-pile
On appelle électrode ou demi-pile le système constitué par un conducteur
électronique en contact avec un conducteur ionique.
Le conducteur électronique est en général un métal et le conducteur ionique une
solution électrolytique (solvant H2O+ électrolyte dissous).
Remarque :
Par extension, on donne le nom d’électrode au conducteur électronique seul.
Lame métallique : conducteur électronique
Solution électrolytique : conducteur ionique
La surface de contact entre les deux conducteurs
est appelée interface, lieu où se produit le transfert
d’électrons entre le métal et la solution.
Fig.1 - Schéma d’une demi-pile
15
IV.2 Potentiel absolu ou tension absolue de l’électrode (fig.2)
Il est égal à la différence des potentiels électriques du métal et de la solution.
Eabs = ΔΦ = Φ (métal) - Φ (solution) .
Fig.2 – Potentiel absolu d’une électrode : Eabs = ∆Φ= Φ(M) - Φ(S)
Electrode 1 : potentiel Φ(M)
Solution : potentiel Φ(S)
Il est impossible de mesurer expérimentalement ce potentiel.
16
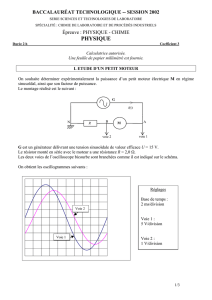
Associons deux demi-piles reliées entre elles par un pont salin (fig.3)
IV.3 Potentiel d’électrode relatif ou tension relative de l’électrode
Fig.3 – Potentiel relatif d’électrode : E = Φ (M2) - Φ (M1) = Eabs2 - Eabs1
Electrode 1 :
potentiel Φ(M1)
Solution 1 :
potentiel Φ(S1)
Electrode 2 :
potentiel Φ(M2)
Solution 2 :
potentiel Φ(S2)
V
E
Pont salin
Ce potentiel d’électrode relatif Eest mesurable. Il est égal à la d.d.p.mesurée aux
bornes de la pile en circuit ouvert.

17
IV.3 Potentiel d’électrode relatif ou tension relative de l’électrode (suite)
Remarque 1 :
Si l’électrode 1 est choisie comme référence, il est alors possible d’établir une
échelle des potentiels relatifs, en prenant pour origine des potentiels celui de
l’électrode de référence.
L’électrode de référence qui a été choisie est l’électrode standard à hydrogène
(ESH) (fig.4) pour laquelle on a fixé Eabs1 = EESH = 0,000V à toute température.
On peut donc écrire, en remplaçant l’électrode 1 de
la fig.3 par l’ESH :
E = Eabs2 - Eabs1 = Eabs2 – EESH = Eabs2 – 0,000 V.
Cette différence est notée [E2]ESH.
Fig.4 – Electrode standard à
hydrogène – EESH = 0,000V
18
En TP, l’électrode de référence au calomel saturée (ECS) est souvent utilisée ; le
potentiel relatif de l’électrode 2 s’écrira alors E = Eabs2 – EECS = [E2]ECS en
remplaçant l’électrode 1 de la fig.3 par l’électrode ECS (fig.5).
Remarque 2 :
On peut exprimer la d.d.p. E entre 2 électrodes
métalliques (cf. fig.3) à l’aide des potentiels relatifs
[Ei]ESH ou [Ei]ECS.
E = [E2]ESH - [E1]ESH = [E2]ECS - [E1]ECS
Fig.5 – Electrode au calomel
saturée [EECS]ESH = 0,244V/ESH
IV.3 Potentiel d’électrode relatif ou tension relative de l’électrode (suite)
19
IV.3 Loi de Nernst
Considérons la pile (fig.6) constituée par l’association d’une électrode ESH et d’une
électrode M mettant en jeu le couple Ox / Red (par exemple, une électrode de
platine plongeant dans une solution contenant les espèces Ox et Red).
Remarque : le réducteur du couple rédox peut jouer le rôle du conducteur
métallique (exemple : électrode d’argent plongeant dans une solution d’ions Ag+).
Electrode 1 = ESH
H+: a(H+) = 1
Electrode 2 = platine
Solution 2 contenant
les espèces Ox et Red.
V
PH2 = 1 bar
H2
Fig.6 – Potentiel rédox du couple Ox / Red
E
Les demi-équations électroniques ayant lieu sur chacune de ces électrodes s’écrivent :
)(2)(
2
1
gaq
HeH ↔+
−+
(2)
)()( aqRaqO RneO
ν
ν
↔+ −
(1)
20
IV.3 Loi de Nernst (suite)
Le potentiel relatif de l’électrode M (appelé aussi potentiel rédox du couple Ox / Red)
est donné par la formule de Nernst :
+==
R
R
O
RORO
a
a
nF
RT
EEE
ν
ν
0
ln
0//
Ce potentiel calculé est égal à la force électromotrice (f.é.m.) algébrique de la pile
associant l’électrode ESH et l’électrode objet de l’étude.
- est le potentiel standard du couple rédox ; c’est une constante à
rechercher dans le tableau 1 (cf. diapo suivante) ;
-Rest la constante des gaz parfaits : R= 8,314 J.K-1.mol-1 ;
-Test la température exprimée en kelvin (K) ;
-Fest la constante de Faraday : F= 96485 C.mol-1 ;
- et représentent respectivement les activités des espèces Ox et Red.
- n représente le nombre d’électrons échangés dans la réaction (2).
0/RO
E
O
a
R
a
 6
7
8
9
10
11
12
13
14
15
16
17
6
7
8
9
10
11
12
13
14
15
16
17
1
/
17
100%