Introduction générale

Introduction générale
Touchant près de 600 nouvelles personnes chaque année en France, la leucémie
myéloïde chronique est une maladie affectant les cellules du sang et de la moelle osseuse
(située au cœur de tous nos os). La moelle osseuse produit l’ensemble des cellules du sang qui
constituent la lignée myéloïde (lexique). Toutes ces cellules proviennent de cellules mères,
appelées cellules souches hématopoïétiques. Dans la leucémie myéloïde chronique (LMC),
toutes les cellules sanguines sont malades. Le terme « chronique » renvoie à la lente évolution
de la maladie.
Aujourd’hui, il est admis que la LMC apparaît à cause de la formation d’un
chromosome (lexique) 22 anormal « Philadelphie » ou Ph, appelé ainsi car découvert dans la
ville de Philadelphie dans les années 60. Ce chromosome entraîne le collage anormal de deux
gènes (lexique).
La mise en évidence de ce chromosome Ph dans les cellules de la moelle osseuse
p
ermet un diagnostic de certitude.
L’évolution de la LMC peut être divisée en 3 phases cliniques distinctes en l’absence de
traitement :
1- Une phase initiale chronique qui correspond le plus souvent à la phase de
diagnostic et caractérisée par un excès de globules blancs (lexique) dans le sang et une
augmentation de la taille de la rate.
2- Une phase accélérée, transition entre la phase chronique et la phase aiguë.
3- La phase de transformation blastique ou de leucémie aiguë (lexique).
Le premier examen que l’on réalise est un hémogramme et/ou un myélogramme.
Des études plus approfondies sont réalisées pour retrouver la présence du chromosome
Philadelphie dans les cellules malades (cytogénétique (lexique) et biologie moléculaire
(lexique)).
La leucémie myéloïde chronique est une maladie sans prédisposition génétique connue
Il existe des facteurs favorisant son apparition, tels que les radiations ou une expositio
n
p
rolongée au benzène, mais en général, on ne connaît pas la cause de l’apparition du
chromosome Ph responsable de la maladie.

Historique de la LMC
La LMC a longtemps été une maladie incurable, jusque dans les années 80. C’est l
a
p
remière leucémie à être décrite en tant que telle. Elle fut caractérisée en 1845 chez deux
p
atients présentant une rate énorme et un sang épais et d’aspect laiteux (dû à un excès de
leucocytes (lexique)), sans qu’on sache expliquer pourquoi.
Vers 1960, de nouvelles techniques se développent et les chercheurs peuvent alors
observer des cellules en division, seul moment où les chromosomes sont visibles dans les
noyaux des cellules. Grâce à ces avancées, Peter Nowell et David Hungerford mettent en
évidence chez les patients présentant les symptômes précédents, un chromosome 22
anormalement court, qu’ils nomment chromosome Philadelphie, du nom de la ville où ils le
découvrirent. Ce chromosome Ph (ou Ph1) fut la première anomalie chromosomique identifiée
dans un cancer.
Ce chromosome est étudié en particulier par Rowley qui montre, en 1973, l’existence
d’un échange de fragments entre les chromosomes 9 et 22 : c’est la translocation t(9;22)
(lexique).
Puis, les connaissances en biologie moléculaire (lexique) évoluant, on montre dans les
années 80 que le chromosome Ph porte un gène anormal, formé de deux morceaux de gènes :
1 fragment du gène BCR sur le chromosome 22, accolé à 1 fragment du gène ABL issu du
chromosome 9. La protéine fabriquée grâce à ce gène BCR-ABL est mutante (lexique) et son
action est responsable des symptômes connus de la maladie.
Différents traitements se développent alors, au fur et à mesure des années : certains
utilisent des molécules chimiques capables de limiter la multiplication des cellules malades
comme l’hydroxyurée (Hydréa), d’autres font appel à une molécule naturelle comme
l’interféron, d’autres encore sont plus radicaux comme la greffe de moelle osseuse qui
remplace les cellules malades par des cellules saines d’un donneur.
Grâce aux connaissances acquises sur le rôle de la protéine BCR-ABL, les années
2000 voient apparaître un traitement révolutionnaire, le Glivec (imatinib mésylate, STI-571)
qui bloque directement la protéine mutante dans les cellules malades et l’empêche d’exercer
son action.
Actuellement, les recherches sur le Glivec continuent pour mieux connaître ses effets à
long terme. Des résistances au traitement apparaissent chez certains patients et déjà, la
seconde génération de traitements bloquants (ou inhibiteurs) est à l’essai.

Diagnostic
Quelques chiffres d’épidémiologie…
On estime qu’il y a 500 à 600 nouveaux cas par an de leucémie myéloïde chronique
(LMC) en France et la fréquence de la maladie est de 1 à 2 cas pour 100 000 habitants.
La LMC peut se déclarer à tout âge, mais le risque d’être atteint augmente avec les
années. L’âge moyen d’apparition de la maladie est d’environ 50 ans, et on note une légère
p
rédominance masculine : 1,4 à 1,5 homme pour 1 femme. On n’a pas découvert de
p
rédisposition génétique pour la LMC.
2,5% des personnes atteintes de LMC ont moins de 10 ans.
15 à 30% des patients ont plus de 60 ans.
Une exposition chronique au benzène ou à des radiations ionisantes peut engendrer
une LMC dite secondaire (5% des cas de LMC).
Symptômes cliniques
La LMC est de plus en plus souvent diagnostiquée par hasard à l’occasion d’une prise
de sang : le taux de globules blancs (leucocytes) est anormalement élevé.
Cependant, le diagnostic peut aussi être réalisé lors d’une consultation pour fatigue
générale, amaigrissement, voire douleurs.
Certaines personnes peuvent être dépistées lors d’un scanner ou d’une échographie, où
l’on détecte une rate de taille augmentée (splénomégalie) caractéristique de la LMC.
La leucémie myéloïde chronique évolue généralement en 3 phases cliniques
successives : une phase chronique pendant laquelle le diagnostic est le plus souvent réalisé,
une phase accélérée et une phase aiguë. Un patient peut cependant être diagnostiqué à
n’importe laquelle de ces 3 phases, et son traitement en dépendra. Toutefois, la très grande
majorité des patients est en phase chronique au moment du diagnostic et l’évolution vers les
p
hases accélérée ou aiguë est bloquée par le traitement.
1- Phase chronique
9 patients sur 10 sont diagnostiqués dans cette phase. Les signes initiaux sont peu
spécifiques :
Amaigrissement, perte de poids
Fatigue générale
Sueurs nocturnes
Parfois, pesanteur du flanc gauche due à la splénomégalie
Lors de la phase chronique, le taux de globules blancs dans le sang devient très élevé
(de 5 000 à 10 000 par mm3, on passe à plus de 50 000 ). Cette augmentation des leucocytes
s’appelle une hyperleucocytose, caractéristique de la LMC.
2- Phase accélérée

C’est une phase de transition entre les phases chronique et aiguë. La maladie s’accélère et les
symptômes deviennent plus évidents :):
Altération de l’état général (asthénie)
Fièvre
Amaigrissement accentué
Sueurs accentuées
Douleurs osseuses
Accentuation de la splénomégalie
La qualité des nouvelles cellules sanguines produites est altérée: les globules rouges
diminuent ainsi que le taux d’hémoglobine (anémie); le nombre de plaquettes diminue aussi.
Le traitement de la phase chronique permet de contrôler le risque de passage en phase
accélérée.
3- Phase aiguë
Aussi appelée phase de transformation blastique, elle survient en l’absence de
traitement. Heureusement, les traitements actuels permettent de stabiliser la phase chronique
de la maladie et évitent le passage en phase aigue.

Techniques d’étude
En dehors de l’hémogramme aujourd’hui automatisé, il existe d’autres techniques
d’étude de la leucémie myéloïde chronique : la cytogénétique et la biologie moléculaire.
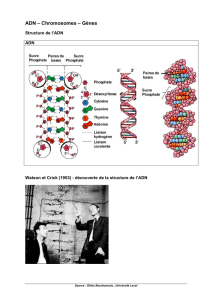
Elles permettent d’étudier les chromosomes et l’ADN, au cœur des cellules.
Au centre du noyau de chaque cellule de l’organisme sont localisés les chromosomes,
constitués d’ADN, c’est notre patrimoine génétique. Grâce à lui, certains ont les yeux bleus,
d’autres les yeux marron, certains ressemblent à leur mère, d’autres à leur père… Tout l’ADN
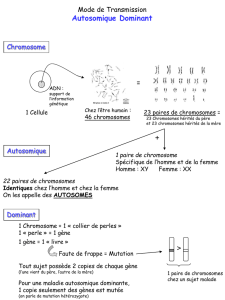
est réparti sur 23 paires de chromosomes chez l’Homme (on possède chaque chromosome en
2 exemplaires: un transmis par notre père + un transmis par notre mère), ce qui fait 46
chromosomes par cellule.
On peut classer ces chromosomes en 23 paires et par taille décroissante : ainsi la paire
de chromosomes 1 contient le chromosome 1 du père et celui de la mère, et le chromosome 1
est le plus grand de tous les chromosomes. De même, la paire 22 est la plus petite. La paire 23
est particulière, ce sont les chromosomes qui déterminent le sexe : 2 chromosomes X pour une
fille, un X et un Y pour un garçon.
Sur un chromosome se succèdent des séquences d’ADN, les gènes. Si on va plus loin,
chaque gène est un patron de fabrication pour une protéine. Si on change, même très peu, la
séquence d’ADN d’un gène, la protéine produite sera différente de celle normalement
produite.
Définition de la translocation
Dans les cellules des personnes atteintes de LMC, un grand morceau du chromosome
22 se casse, un petit morceau du chromosome 9 se casse et les deux morceaux s’échangent et
se recollent : le petit morceau du 9 se colle sur le 22 cassé, et inversement, le grand fragment
du 22 va s’associer au 9 cassé. Cet échange de fragments de chromosomes s’appelle une
translocation, ici entre les chromosomes 9 et 22. Ce chromosome 22 qui contient un morceau
du 9 est aussi appelé chromosome Philadelphie.
Au niveau de l’ADN, le gène Abelson (ABL) est cassé en deux sur le chromosome 9,
ainsi que le gène BCR sur le chromosome 22. Quand les morceaux de chromosomes
s’échangent, le fragment cassé du gène BCR va se coller au morceau restant du gène ABL sur
le chromosome 9, et de même, le morceau cassé du gène ABL va se coller au fragment restant
du gène BCR sur le chromosome 22. On obtient donc sur le chromosome 22 un gène
anormal BCR-ABL formé du début du gène BCR et de la fin du gène ABL, et sur le
chromosome 9 un gène anormal ABL-BCR, composé du début du gène ABL et de la fin du
gène BCR.
Le gène muté BCR-ABL peut être détecté, ainsi que la protéine dont il permet la
fabrication.
 6
7
8
9
10
11
12
6
7
8
9
10
11
12
1
/
12
100%