1 - Constellation (UQAC)

UNIVERSITE DU QUÉBEC
MEMOIRE
PRÉSENTÉ À
L'UNIVERSITÉ DU QUÉBEC À CHICOUTIMI
COMME EXIGENCE PARTIELLE
DE LA MAÎTRISE EN RESSOURCES RENOUVELABLES
PAR
FLEUR GAGNON
B.Sc (CHIMIE)
FORMATION DE GLYCOSIDE
À PARTIR DE
L'ACIDE
BETULINIQUE
AOUT 2004

bibliothèque
Paul-Emile-Bouletj
UIUQAC
Mise en garde/Advice
Afin de rendre accessible au plus
grand nombre le résultat des
travaux de recherche menés par ses
étudiants gradués et dans l'esprit des
règles qui régissent le dépôt et la
diffusion des mémoires et thèses
produits dans cette Institution,
l'Université du Québec à
Chicoutimi (UQAC) est fière de
rendre accessible une version
complète et gratuite de cette œuvre.
Motivated by a desire to make the
results of its graduate students'
research accessible to all, and in
accordance with the rules
governing the acceptation and
diffusion of dissertations and
theses in this Institution, the
Université du Québec à
Chicoutimi (UQAC) is proud to
make a complete version of this
work available at no cost to the
reader.
L'auteur conserve néanmoins la
propriété du droit d'auteur qui
protège ce mémoire ou cette thèse.
Ni le mémoire ou la thèse ni des
extraits substantiels de ceux-ci ne
peuvent être imprimés ou autrement
reproduits sans son autorisation.
The author retains ownership of the
copyright of this dissertation or
thesis.
Neither the dissertation or
thesis,
nor substantial extracts from
it, may be printed or otherwise
reproduced without the author's
permission.

À mes parents, Jean-Pierre et Mercedes,
qui m'ont inculqué le goût du savoir.

Ill
RESUME
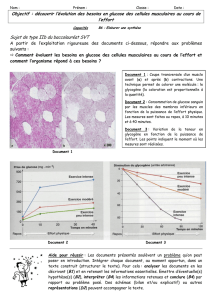
L'acide bétulinique 1, un triterpène au squelette de lupane, est connu pour ses
activités thérapeutiques. Entre autres, ce composé possède une activité anticancéreuse et
antivirale. L'acide bétulinique 1.
s'est
avéré cytotoxique contre de nombreuses lignées de
cellules cancéreuses. De plus, il présente une faible toxicité pour les cellules saines de
l'organisme ce qui en fait un traitement potentiel pour certaines tumeurs cancéreuses.
Cependant, l'acide bétulinique 1 présente une limite d'application étant donné sa faible
biodisponibilité dans l'organisme. La faible biodisponibilité du composé est possiblement
due à sa faible hydrophilicité.
Ce projet vise la synthèse d'un analogue glycosidique de l'acide bétulinique
1^
pour en
augmenter l'hydrosolubilité dans le but d'améliorer la biodisponibilité du composé dans
l'organisme tout en conservant une activité anticancéreuse. Pour ce faire, une section
polaire et hydrophile, le glucose 4, est ajoutée à la fonction alcool du composé. L'objectif
principal du projet de recherche est l'élaboration de la synthèse d'un dérivé glycosidique
bioactif de l'acide bétulinique
1.
pour en augmenter l'hydrosolubilité.
La formation de l'acide 3-Op-D-glucopyranosylbétulinique 15 a été réalisée en cinq
étapes en utilisant l'activation par les trihalogénoacétimidates. Le couplage de l'acide
bétulinique 1 et du l-trichloroacétimidates-2,3,4,6-tétra-O-benzoyl-glucopyranose 7b a été

IV
effectuée à l'aide du catalyseur triméthylsilyl trifluorométhanesulfonate (TMSOTf). La
synthèse a été réalisée avec un rendement de 32 %.
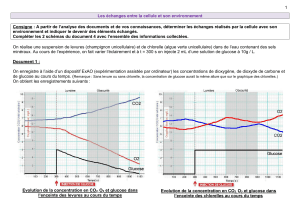
Le coefficient de partage de l'acide 3-0-(3-D-glucopyranosylbétulmique 15 a été
évalué et montre que l'hydrophilicité de ce dernier est beaucoup plus importante que celle
du composé de départ, l'acide bétulinique 1, ce qui pourrait permettre d'améliorer sa
biodisponibilité in vivo.
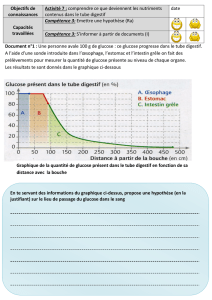
L'ajout d'une section sucre à l'acide bétulinique 1. peut avoir une influence sur
l'activité anticancéreuse de la molécule. L'activité anticancéreuse de l'acide 3-0-p-D-
glucopyranosylbétulinique 15 a été évaluée in vitro envers deux lignées de cellules
cancéreuses humaines. L'acide 3-O-p-D-glucopyranosylbétulinique 15 possède une activité
anticancéreuse envers les deux lignées de cellules cancéreuses à l'étude avec une
concentration qui inhibe la croissance de 50% des cellules (IC50) de 12,73 ug/ml envers les
cellules du cancer du poumon (A549) et de 25,13 jxg/ml envers celles du cancer du colon.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
1
/
180
100%