TP 2 : Minimisation d`énergie - DSIMB

M1 BI Université Paris Diderot – Paris 7
33GB4131 Modélisation moléculaire
TP 2 : Minimisation d'énergie
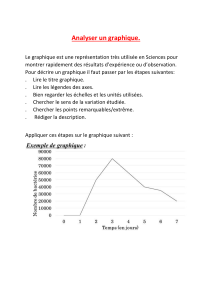
But : Afin de vous donner un aperçu des différentes techniques de minimisation, nous allons dans un
premier temps regarder les résultats sur un petit système constitué de deux molécules d'eau (comme
par hasard...). Dans un deuxième temps, on observera comment se comportent les mêmes algorithmes
sur une petite protéine, la Barstar. Tous ces résultats ont été générés avec le programme et le champ
de forces de CHARMM.
1) Minimisation de deux molécules d’eau.
Pour cet exercice, vous allez utiliser rasmol (ou pymol) pour visualiser le système des deux molécules
d'eau, ainsi que xmgrace pour regarder des graphes. Voici des sites d'aide :
http://www.bernstein-plus-sons.com/software/rasmol/doc/rasmol.html
http://pymol.sourceforge.net/newman/user/toc.html
http://plasma-gate.weizmann.ac.il/Grace/doc/UsersGuide.html
a) Calcul d'énergie.
•Créer un nouveau répertoire mini_water dans lequel vous ferez cette partie du TP.
•Récupérer le fichier water.pdb et enregistrer le dans le répertoire. (tous les fichiers sont
disponibles à la page : http://www.dsimb.inserm.fr/~fuchs/M1BI/index.html
•Regarder le système avec rasmol. L'orientation des molécules d'eau vous semble-t-elle
favorable ? Mesurer la distance O-O avec la commande set picking distance. À
quel signe vous attendez-vous pour l'énergie électrostatique ?
•Regarder le résultat du calcul d'énergie sur la page web. Si on minimisait le système, à
quelle(s) modification(s) vous attendriez vous au niveau des termes d'énergie et de la
géométrie ?
b) Minimisation.
Nous avons lancé une minimisation pour vous en utilisant 4 algorithmes disponibles dans le
programme CHARMM : Steepest Descent, Gradient Conjugué, ABNR (Adopted-Basis Newton-
Raphson) et Newton-Raphson. Dans les 4 cas, nous avons minimisé jusqu'à ce que le RMS du
gradient de l'énergie (GRMS dans les fichiers CHARMM) soit inférieur à 10-4 kcal.mol-1.Å-1. Un
maximum de 10000 pas de minimisation a été imposé au cas où la méthode n'aurait pas convergé.
•Pour l'algorithme Steepest Descent, regarder le fichier de sortie CHARMM ainsi que le
graphe avec le programme xmgrace (enregistrer chaque graphe sur le disque puis lancer
xmgrace de la manière suivante xmgrace fichier.xvg). Que constatez-vous ?
L'algorithme vous semble-t-il efficace ?
Pour xmgrace : en haut à gauche vous avez un bouton avec une loupe permettant de faire un zoom
avec la souris, pour revenir au graphe complet, cliquer sur le bouton As). N'hésitez pas à zoomer
sur les diverses zones du graphe.
Pour le fichier de sortie de CHARMM : il contient un bilan des énergies le long des pas de
minimisation (toutes les lignes commençant par MINI>, MINI INTERN>).
•Regarder de même les fichiers de sortie et les graphes pour le Gradient Conjugué, l'ABNR et
Newton-Raphson. Les 4 algorithmes convergent-ils vers la même énergie ?
•Regarder les 4 structures minimisées. Sont-elles différentes ? Est-ce que cela vous paraît
cohérent par rapport à la différence d'énergie ?
•Regarder le graphe de comparaison des 4 algorithmes. Quelles sont les méthodes qui ont ou
n'ont pas convergé ? Quelle est selon vous la plus efficace ? Pouvez-vous expliquer
pourquoi ?
2) Minimisation d'une petite protéine, la barstar.
1/2 2008-10-22

M1 BI Université Paris Diderot – Paris 7
33GB4131 Modélisation moléculaire
Comme dans l’exercice précédent, nous allons comparer les différents algorithmes de minimisation
disponibles dans le programme CHARMM, mais cette fois-ci sur la protéine barstar (code PDB :
1BTA) qui est un inhibiteur de ribonucléase.
•Comme dans le 1), observer le comportement de chaque algorithme de minimisation en
utilisant les fichiers sur la page web. Pour chaque algorithme, noter l’énergie finale (bien
observer les différentes contributions), le nombre de pas pour converger ou l’absence de
convergence, le RMS du gradient de l’énergie (GRMS), le temps CPU.
•Regarder la structure après chaque minimisation (vous pouvez les comparer avec pymol) et
comparer la avec la structure de départ en mode ruban, puis en mode ‘wireframe’. Observez-
vous des différences importantes.
•Quel algorithme vous semble le plus efficace ? S’agit-il du même que dans le 1) ? Comment
expliquez-vous ce résultat ?
Conseil : vous pouvez établir un tableau récapitulatif des différentes méthodes de minimisation pour
chaque système étudié (2 molécules H2O et barstar) :
SD CG ABNR NRAP
Epot finale
Eelec finale
EVDW finale
Eliée finale
Convergence ?
Nb de pas pour
converger
GRMS final
elapsed time
Observation
Structure
minimisée
2/2 2008-10-22
1
/
2
100%