Obésités génétiques et syndromiques

Obésités génétiques et
syndromiques
3ième symposium de la Division d’endocrinologie,
diabétologie et obésité pédiatrique, CHUV Lausanne
Pr Pascal Barat
Endocrinologie pédiatrique
CHU Bordeaux , France
REPPOP Aquitaine

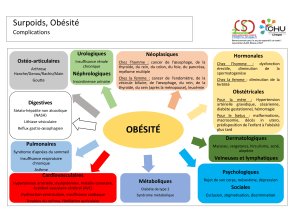
Classification de l’obésité
pédiatrique
•Obésité commune
•Obésités endocriniennes
–Sd de Cushing
–Hypothyroïdie
•Obésités monogéniques
•Obésités syndromiques

Plan
• Part de la génétique dans l’obésité
commune
•Obésités monogéniques
•Obésité syndromiques

Part de la génétique dans
l’explication de l’obésité commune

 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
1
/
51
100%