Cardiopathies congénitales de l`adulte

Dossier – Cardiopathies congénitales
Cardiopathies congénitales de l’adulte :
conduite diagnostique
Laurence Iserin
1,2
, Magalie Ladouceur
1
1
Unité de cardiopathie congénitale de l’adulte, Pôle cardiovasculaire, hôpital européen Georges Pompidou, 15 rue Leblanc, 75015 Paris
2
Service de cardiologie pédiatrique, hôpital Necker Enfants Malades, Paris
Résumé.Le diagnostic de cardiopathie congénitale native à l’âge adulte n’est pas inhabituel, et est en général assuré par l’échographie
transthoracique, qui fait partie de la consultation de cardiologie congénitale, ou par l’échographie transœsophagienne. Il peut s’agir de lésions
simples, comme la communication interauriculaire (CIA), le canal artériel ou la sténose pulmonaire, ou plus complexes, comme la maladie
d’Ebstein ou la double discordance. Dans tous les cas, ces patients, qui peuvent cumuler pathologies congénitales et acquises (hypertension
artérielle, parfois maladie coronaire, troubles du rythme et évolutivité des lésions artérielles pulmonaires), sont à haut risque de complications,
médicales comme chirurgicales. Le bilan préthérapeutique plus complet des lésions fait souvent appel à d’autres techniques (scanner et IRM).
Le cathétérisme cardiaque diagnostique a une part peu importante en dehors de l’évaluation des résistances pulmonaires en cas d’hypertension
artérielle pulmonaire. Le cathétérisme interventionnel se développe pour les lésions simples (fermeture de CIA, de canal artériel ou dilatation
au ballon des sténoses pulmonaires). Cependant, la plupart des patients ont déjà été opérés dans l’enfance. Le diagnostic de la cardiopathie
initiale est rarement méconnu, le diagnostic des complications peut être difficile chez des patients complexes opérés par des techniques parfois
anciennes (fuite pulmonaire résiduelle des tétralogies de Fallot, dysfonction des ventricules droits systémiques). Environ 50 % des patients
devront avoir un suivi régulier car un certain nombre de patients auront besoin d’une réopération à l’âge adulte, ou d’une procédure
interventionnelle de cathétérisme ou de rythmologie.
Mots clés : CIA, cathétérisme interventionel, sténose pulmonaire
Abstract. Diagnosis of congenital heart disease. Diagnosis of congenital heart disease (CHD) in adulthood is not unusual. Adult CHD
is often suspected by clinical examination and confirmed by echocardiography (which is part of the clinic for adult patients with congenital heart
defects). Lesions can be simple, such as atrial septal defect, arterial duct, native coarctation or pulmonary stenosis. The patient can also survive
into adulthood with a complex defect as Ebstein disease or double discordance. TOE can reliably assess intra cardiac defects. In adults complete
assessment is generally achieved with MRI or CT scanners in experienced centers. Use of cardiac catheters for diagnosis is mainly for pulmonary
resistance assessment. Interventional catheterism is an increasingly used technique (ASD or duct closure, balloon dilatation of pulmonary
stenosis). The managment of these patients is frequently complex because of the evolution of native disease (calcifications of abnormal
congenital valves, arrythmias, pulmonary artery disease), or because of acquired cardiac pathology as systemic hypertension, and sometimes
coronary artery disease. The majority of patients with CHD have been operated on in infancy. Initial diagnostics are often known, but sequellae
and complications can be specific. About 50% of these patients require careful follow up, as some of them will need further operations,
interventional catheterism or interventional antiarrythmic procedures in adulthood.
Key words: atrial septal defect, interventional catheterism, pulmonary stenosis
Bases épidémiologiques
L’incidence des cardiopathies
congénitales (CC) est d’environ de 5 à
8 pour mille naissances vivantes.
Avant l’avènement de la chirurgie car-
diaque à cœur ouvert, 60 % des en-
fants atteints de cardiopathies congé-
nitales mourraient. De nos jours, 70 à
80 % atteignent l’adolescence et l’âge
adulte et on estime leur nombre aux
États-Unis à plus d’un million [1]. La
majorité des patients ayant une car-
diopathie congénitale bénéficient ac-
tuellement d’une cure chirurgicale
dans la première année de vie, ce qui
ne signifie pas qu’ils sont tous guéris
car certains posent des problèmes spé-
cifiques, dont le diagnostic est parfois
difficile par les méthodes échographi-
ques simples. D’autre part, beaucoup
de patients atteignent l’âge adulte sans
recours à la chirurgie, que la cardio-
pathie soit bien tolérée et parfois
même découverte à l’âge adulte, ou
qu’elle soit irréparable et alors le plus
souvent cyanogène.
Le problème diagnostique se pose
donc devant une CC de découverte
tardive jamais opérée, ou au contraire
doi: 10.1684/mtc.2007.0087
m
t
c
Tirés à part : L. Iserin
mt cardio 2007 ; 3 (2) : 93-101
mt cardio, vol. 3, n° 2, mars-avril 2007 93
Dossier – Cardiopathies congénitales
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

devant une CC opérée dans l’enfance (avec un diagnostic
initial parfois même méconnu par le patient) où il s’agit
d’identifier les séquelles et complications de cette chirur-
gie à l’âge adulte.
Cardiopathies non opérées
à l’âge adulte
Il est impossible d’envisager ici l’évolution spontanée
de toutes les CC et nous détaillerons plus particulièrement
ici celles qui sont relativement fréquentes et dont la survie
est attendue à l’âge adulte. Elles peuvent être réparties en
deux groupes :
–anomalies bien tolérées qui peuvent être découver-
tes à l’âge adulte : bicuspidie aortique, coarctation, com-
munication interauriculaire (CIA), canal artériel, commu-
nication interventriculaire (CIV), sténose pulmonaire,
anomalie d’Ebstein, double discordance ;
–malformations inopérables, en général complexes,
cyanogènes et connues depuis l’enfance.
Cardiopathies pouvant être découvertes
à l’âge adulte
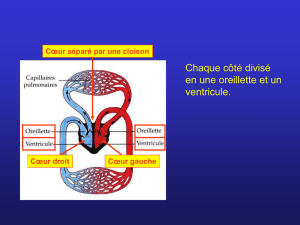
Communications interauriculaires
Les CIA (surtout de type ostium secundum, situées au
centre du septum interauriculaire) sont très fréquemment
découvertes tardivement en raison de l’absence de symp-
tômes jusqu’à un âge avancé et de signes physiques dis-
crets. En cas de défaut large, la plupart des patients sont
symptomatiques à partir de 50 ans : des troubles du
rythme à type de flutter et de fibrillation auriculaire appa-
raissent, l’hypertension artérielle pulmonaire, même mo-
dérée, impose une surcharge de travail au ventricule
droit ; une insuffisance mitrale peut aussi survenir. Le dia-
gnostic de CIA doit être évoqué en échographie devant une
dilatation des cavités droites. Celle-ci est due à la surcharge
volémique créée par le shunt gauche-droit auriculaire.
La présence d’un défect du septum interauriculaire par voie
sous-costale permet de porter le diagnostic. Il faut essayer
de dégager un plan sagittal permettant de voir l’abouche-
ment à la fois de la veine cave inférieure et supérieure, en
particulier pour la CIA sinus venosus
. Lorsque le septum
interauriculaire n’est pas vu dans cette incidence, en
particulier chez les patients obèses ou longilignes, le
diagnostic de CIA ne peut être porté qu’en échographie
transœsophagienne. Cet examen permet de mieux étudier
la région de la fosse ovale et le rapport entre le septum
interauriculaire et la veine cave supérieure, et de
voir l’abouchement à l’oreillette gauche des veines
pulmonaires.
L’estimation des pressions pulmonaires se fait sur l’IT
(qui surestime souvent la PAPs en raison d’un gradient de
débit sur la voie pulmonaire) et sur l’IP. Le calcul du
QP/QS est possible, mais entaché d’erreur chez l’adulte en
raison de la difficulté à mesurer l’anneau pulmonaire.
L’échographie tridimensionnelle permet de mieux étudier
la forme et les rapports anatomiques des CIA.
Les shunts significatifs entraînant une dilatation impor-
tante des cavités droites (taille du ventricule droit au moins
égale à celle du gauche) doivent donc être supprimés
auparavant.
La survenue d’une hypertension artérielle pulmonaire
(HTAP) fixée est rare (< 8 % des CIA ostium secundum et
plus fréquente dans le cas des sinus venosus) mais plus
fréquente chez les femmes et favorisée par les grossesses
multiples. Elle est évoquée sur le plan clinique par une
désaturation d’effort, une gêne fonctionnelle importante,
et à la radiographie thoracique par l’absence de surcharge
vasculaire pulmonaire avec plutôt un aspect de dilatation
proximale des branches et une raréfaction des vaisseaux
en périphérie. En échographie, la pression artérielle pul-
monaire (PAP) systolique dépasse 50 mm. La PAP diasto-
lique est élevée, de même que les résistances pulmonai-
res, avec peu de débit sur la voie pulmonaire voire une
inversion du shunt à travers la CIA [2].
L’HTAP modérée avec hyperdébit pulmonaire est fré-
quente et progresse avec l’âge (POG augmentant).
En l’absence de CIA détectée, un retour veineux pul-
monaire anormal doit être recherché (par une ETO ou plus
facilement un scanner ou une IRM) (figures 1-3).
Canal artériel
Le canal artériel relie l’aorte descendante (isthme) à
l’artère pulmonaire gauche. Il est responsable d’une sur-
charge volumique de l’OG et du ventricule gauche et doit
donc être recherché devant une dilatation des cavités
gauches. Il est difficilement visible chez l’adulte en 2D.
On recueille surtout un jet ascendant dans l’artère pulmo-
naire gauche en Doppler couleur et/ou en Doppler
continu en parasternal petit axe. Il s’agit d’un flux continu
avec une vitesse maximale enregistrée en systole. La vi-
tesse maximale alors mesurée permet d’évaluer la PAPs
Liste des abréviations
CC : cardiopathie congénitale
CIA : communication interauriculaire
CIV : communication interventriculaire
DTI : Doppler tissue imaging
ETO : échocardiographie transœsophagienne
HTA : hypertension artérielle
HTAP : hypertension artérielle pulmonaire
IP : insuffisance pulmonaire
IRM : imagerie par résonance magnétique
IT : insuffisance tricuspide
OG : oreillette gauche
PAP : pression artérielle pulmonaire
PAPs : pression artérielle pulmonaire systolique
POG : pression dans l’oreillette gauche
Diagnostic des cardiopathies congénitales de l’adulte
mt cardio, vol. 3, n° 2, mars-avril 2007
94
Dossier – Cardiopathies congénitales
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

(gradient systolodiastolique aorte-AP) (figure 4). En cas de
canal artériel de très petite taille et asymptomatique le plus
souvent, l’échographie permet de faire le diagnostic par la
découverte souvent fortuite de ce flux couleur. Si le canal
artériel est de calibre modeste, les patients sont asympto-
matiques jusqu’à l’âge de 30 ans ou plus mais une insuf-
fisance cardiaque, souvent annoncée par une fibrillation
auriculaire et associée à une insuffisance pulmonaire et
une dilatation de l’artère pulmonaire, peut finir par appa-
raître. Un vol coronaire est possible, puisque le shunt du
canal artériel affecte systole et diastole.
Si le vaisseau est très large, il peut entraîner une HTAP
fixée, entraînant un shunt bidirectionnel à travers le canal
et une désaturation préférentielle aux membres inférieurs.
Le diagnostic est alors clinique car en échographie, la
vélocité du canal étant très faible, il est très difficile de voir
le canal artériel. Le diagnostic d’HTAP peut être fait en
méconnaissant le diagnostic de canal artériel.
Coarctation aortique
La découverte tardive d’une coarctation aortique, en
général lors du bilan d’une HTA n’est pas rare. Le diagnos-
tic est clinique : asymétrie à la palpation des pouls entre
AB
V
5
10
15
Figure 1.A)CIAostium secundum avec shunt gauche-droit au Doppler couleur en 4 cavités. B) CIA large extension postérieure avec HTAP (VD
dilaté et hypertrophié).
Figure 2.CIA résiduelle de type ostium primum. La CIA a déjà été opérée avec un patch calcifié et le défect est au niveau des valves
auriculoventriculaires.
mt cardio, vol. 3, n° 2, mars-avril 2007 95
Dossier – Cardiopathies congénitales
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

les membres supérieurs et inférieurs, ou asymétrie tension-
nelle. La longueur de l’évolution favorise le développe-
ment d’une importante circulation collatérale, des lésions
intimales des artères systémiques, d’où risques de dissec-
tion aortique, de rupture d’anévrisme dans le polygone de
Willis ou de maladie coronaire précoce. La moyenne de
survie spontanée de ces patients est de 35 ans en raison
des complications de l’HTA. De plus, l’association à une
bicuspidie aortique (un tiers des cas), augmente le risque
d’endocardite et de dilatation dangereuse de l’aorte as-
cendante. Dix à 20 % des coarctations aortiques seraient
diagnostiquées à l’adolescence ou à l’âge adulte [3]. La
recherche d’une coarctation doit être systématique chez le
sujet jeune hypertendu. En échographie, elle se recherche
en incidence suprasternale. Chez l’adulte, il est difficile de
voir la morphologie de la sténose. C’est au Doppler cou-
leur et continu que l’on peut porter le diagnostic. Le
Doppler couleur montre un flux pulsatile dans l’aorte
sus-stricturale et un flux « mosaïque » dans le rétrécisse-
ment de l’isthme et le Doppler continu enregistre un flux
accéléré à double enveloppe spectrale, l’une correspon-
dant au flux sus-strictural et l’autre au flux trans-sténotique
(figure 5). La sévérité de la coarctation dépendra plus de la
présence d’un prolongement diastolique du flux antéro-
grade que du gradient systolique mesuré qui surestime la
sténose (figure 3). L’IRM est largement supérieure à
l’échographie pour faire le bilan de cette malformation
(morphologie de la sténose et de l’aorte sus- et sous-
stricturale, collatéralité).
Sténose valvulaire pulmonaire
Une sténose valvulaire pulmonaire est souvent décou-
verte devant un souffle systolique, et parfois méconnue
(considérée comme un souffle anorganique). La valve peut
se calcifier avec le temps, l’obstacle étant progressif ; le
ventricule droit est longtemps adapté, hypertrophique et
finalement une cyanose par inversion d’un shunt atrial
peut majorer les symptômes d’effort, alors que la dé-
faillance droite est rare et apparaît quand la fuite tricus-
pide est importante. Une sténose musculaire infundibu-
laire réactionnelle est souvent associée et peut ne
régresser que partiellement après dilatation au ballon.
L’échographie fait le diagnostic et permet de mesurer
l’anneau pour la valvuloplastie percutanée.
Maladie d’Ebstein
La maladie d’Ebstein est définie par l’accolement du
feuillet septal et parfois postérieur de la tricuspide le long
de la paroi du ventricule droit, ce qui délimite une cham-
bre intermédiaire entre l’OD et le ventricule droit. Malgré
une cardiomégalie importante, le patient peut être long-
temps asymptomatique et la maladie ne se révéler que par
une arythmie auriculaire et/ou supraventriculaire avec
syndrome de Wolff-Parkinson-White à l’âge adulte.
Figure 3.CIA sinus venosus en ETO. La VCS est à cheval sur le défect
haut situé.
V
5
V
5
10
15
10
15
Figure 4.Flux couleur de canal artériel avec flux Doppler continu mais gradient faible témoignant d’une HTAP.
Diagnostic des cardiopathies congénitales de l’adulte
mt cardio, vol. 3, n° 2, mars-avril 2007
96
Dossier – Cardiopathies congénitales
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

Les patients peuvent être également dyspnéiques et la
dyspnée est liée au degré de cyanose par shunt droite-
gauche auriculaire à travers le foramen ovale. La survie
actuarielle pour les patients nés vivants est de 67 % à un
an et de 59 % à 10 ans [4]. La plastie chirurgicale mobilise
le feuillet valvulaire antérieur, et peut souvent chez
l’adulte comporter une dérivation cavopulmonaire par-
tielle. L’importance de la fuite tricuspide, l’évaluation de
la fonction du ventricule droit, la taille du ventricule droit
effectif sont des éléments fondamentaux dans la descrip-
tion échographique de cette pathologie, qui doit être
complétée au mieux par une IRM.
Doubles discordances
Dans l’enfance, la cardiopathie est souvent décou-
verte, surtout quand une anomalie y est associée (fuite
tricuspide par malformation de la valve tricuspide, CIV,
sténose pulmonaire ou sous-pulmonaire). Des anomalies
de conduction du type bloc auriculoventriculaire complet
peuvent également faire découvrir la pathologie. La survie
à l’âge adulte parfois avancé est fréquente en l’absence de
malformations associées. Elle peut être découverte de
façon fortuite à l’échographie cardiaque ou devant des
troubles conductifs. L’échographie, avec une l’analyse pas
à pas de l’anatomie, permet en général de bien décrire la
cardiopathie. Comme pour toutes les CC, elle doit s’ap-
puyer sur une analyse segmentaire (analyse du situs, ana-
lyse des connexions atrioventriculaires, puis des
connexions ventriculo-artérielles, et de la position des
ventricules et des vaisseaux entre eux). L’aspect le plus
commun est celui en situs solitus (normal) avec lévocar-
die. L’aspect qui doit attirer l’attention est la présence d’un
ventricule situé à gauche de morphologie particulière. Il
présente les caractéristiques d’un ventricule droit morpho-
logique, à savoir des trabéculations grossières (figure 6),la
présence d’une bandelette modératrice, la présence d’un
infundibulum (discontinuité entre la valve d’entrée, tricus-
pide et la valve de sortie ici aortique), et une valve d’entrée
tricuspide. En incidence apicale 4 cavités, le ventricule
situé à gauche est plus trabéculé, et sa valve auriculoven-
triculaire s’insère de façon plus apicale que la valve située
à droite. Le ventricule droit situé à gauche donne nais-
sance à l’aorte. Les vaisseaux sont L-malposés entre eux,
c’est-à-dire que l’aorte est antérieure et à gauche de l’ar-
tère pulmonaire (figure 7). On voit naître l’artère pulmo-
naire du ventricule gauche située à droite en apical cinq
cavités. L’évaluation de la fonction ventriculaire droite
systémique est difficile. En effet, la géométrie du ventricule
droit étant différente de celle du ventricule gauche, les
mesures comme la FEVG par la méthode de Simpson ou la
fraction de raccourcissement des parois du ventricule
droit, ne sont pas des méthodes validées. De plus, les
V
V
5
10
15
5
10
15
Figure 5.Coarctation modérée mais flux couleur en faveur d’une sténose isthmique avec flux Doppler et prolongement diastolique.
V
5
10
Figure 6.Double discordance : la valve auriculoventriculaire gauche
(tricuspide) s’insère sur le septum et de façon plus apicale que la
valve auriculoventriculaire droite.
mt cardio, vol. 3, n° 2, mars-avril 2007 97
Dossier – Cardiopathies congénitales
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.
 6
7
8
9
6
7
8
9
1
/
9
100%