Fièvre méditerranéenne familiale

1. Introduction
La maladie périodique, ou fièvre méditerranéenne familiale
(FMF, MIM 249100), est une affection génétique se trans-
mettant selon un mode autosomique récessif,et caractérisée
par des épisodes récurrents de fièvre accompagnés de
signes cliniques en rapport avec une inflammation des
séreuses. Cette maladie touche essentiellement les popula-
tions du pourtour méditerranéen, en particulier les popu-
lations arméniennes, turques, juives séfarades, et arabes,
dans lesquelles l’estimation de la fréquence des hétérozy-
gotes pour une mutation du gène en cause varie de 1/6 à
1/20 . La gravité de cette affection réside essentiellement
dans le risque de survenue d’une amylose généralisée dont
la traduction clinique la plus sévère est une atteinte rénale
progressive qui,à terme, évolue vers une insuffisance rénale.
Le traitement de cette affection repose sur la prise régulière
de colchicine (1 à 2 mg par jour). Une étude menée chez
350 enfants atteints de FMF traités au long terme par la col-
chicine, rapporte une rémission totale ou partielle des
crises douloureuses chez respectivement 64 % et 31 % des
patients (1). Quel que soit le type de réponse des patients
au moment des crises, il a été montré que ce traitement de
fond prévenait le développement d’une amylose rénale.
Fièvre
méditerranéenne
familiale
Commission « Pratique de la Génétique »
iche de synthèse des données scientifiques
utiles au Conseil Génétique

2. Diagnostic du phénotype
2.1. Éléments du diagnostic positif
Devant l’absence de critère objectif de dia-
gnostic, le diagnostic de FMF est porté sur un
faisceau d’arguments cliniques non spéci-
fiques. Les accès aigus sont une des caracté-
ristiques essentielles de la maladie ; ils débu-
tent brutalement, durent un à trois jours, se
répètent sans période, de façon imprévisible.
Ils associent, de façon inconstante une fièvre,
des douleurs abdominales (manifestation la
plus caractéristique), articulaires (arthrite
aseptique),ou thoraciques (pleurésie),ou des
signes cutanés (pseudo-érysipèle), et un syn-
drome biologique inflammatoire non spéci-
fique.
Différents critères ont été proposés : Livneh
et al. (2) ont, dans une étude rétrospective,
évalué des critères « simplifiés » et proposent
de poser le diagnostic de FMF sur l’observa-
tion d’au moins trois crises récurrentes du
même type présentant au moins un critère
majeur (péritonite généralisée, pleurésie,
arthrite localisée, fièvre ou atteinte abdomi-
nale incomplète), ou au moins deux critères
mineurs (atteinte incomplète impliquant au
moins le thorax ou une articulation, douleur
à l’effort, réponse favorable à la colchicine).
Des critères plus restrictifs dits critères de Tel
Hashomer, ont été proposés par Pras (3). Il
s’agit dans tous les cas d’un diagnostic d’éli-
mination qui nécessite parfois la réalisation
d’explorations invasives (laparotomies,
arthrotomies...). Il est pourtant essentiel
d’assurer le diagnostic de cette affection,
compte tenu de l’efficacité thérapeutique de
la colchicine qui, prise quotidiennement et
au long cours,permet de prévenir la survenue
d’une atteinte rénale (efficacité voisine de
100 %), et, dans la majeure partie des cas, de
réduire la fréquence et l’intensité des crises.
Enfin, citons la description de rares cas de
FMF révélés par une amylose rénale inaugu-
rale.
2.2. Diagnostic différentiel
Le diagnostic de FMF étant un diagnostic
d’élimination, un grand nombre d’affections
peut mimer une FMF.Soulignons par ailleurs
l’existence d’autres maladies dites pério
diques :
le syndrome Hyperimmunoglobulinémie
D avec
fièvre périodique (MIM 260920), la fièvre
familiale hibernienne (MIM 142680), et le
syndrome de Marshall (ou PFAPA, Periodic
Fever, Aphtous stomatitis, Pharyngitis and
Adenitis).
3. Génétique
3.1. Mode de transmission
Autosomique récessif
Un mode de transmission pseudo-dominant
a été rapporté dans plusieurs familles (4/5) ;
ceci s’explique par la grande fréquence des
porteurs hétérozygotes d’une mutation du
gène MEFV dans les populations à risque
(1/6 à 1/20), la probabilité d’une union entre
un patient atteint de FMF et un individu
hétérozygote étant donc non négligeable.
Les études moléculaires récentes ont par
ailleurs démontré une pénétrance incomplète
du phénotype clinique pathologique chez des
individus porteurs de deux allèles MEFV
mutés (4/5).
3.2. Locus
16p13 : gène MEFV
Ce locus a cependant été exclu dans deux
familles turques (6).
3.3. Gène
Le gène MEFV (MEditerranean FeVer),locali-
sé sur le bras court du chromosome 16,com-
porte 10 exons couvrant environ 14 kb
d’ADN génomique (7/8).
L’ADN complémentaire correspondant de
3,7 kb coderait une protéine de 781 acides
aminés appelée marenostrine ou pyrine. Bien
que la fonction de cette protéine soit à ce jour
totalement inconnue, l’analyse de la structure
primaire suggère qu’il s’agit d’un facteur de
transcription dont l’ARN est spécifiquement
exprimé dans les polynucléaires neutrophiles
et les monocytes. Cette hypothèse a récem-
ment été confortée par la description d’une
nouvelle isoforme, délétée de l’exon 2 (géné-
rée par épissage alternatif) et qui, selon des
études menées in vitro,donne naissance à une
protéine nucléaire (9).
Fièvre
méditerranéenne
familiale

3.4. Mutations
À ce jour, plus d’une vingtaine de mutations
faux-sens ont été identifiées. Les plus fré-
quentes sont situées dans l’exon 10 du gène.
Plusieurs allèles complexes associant deux ou
trois variations de séquence ont également
été identifiés. Ces mutations ponctuelles
n’ont encore fait l’objet d’aucune étude fonc-
tionnelle. De nombreux polymorphismes
répartis tout le long du gène ont été décrits.
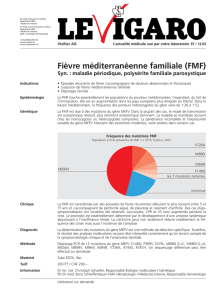
Liste des principales mutations identifiées
Exon 10
M694V (nt 2080 ATG->GTG)
V726A (nt 2177 GTT->GCT)
M680I (nt 2040 ATG->ATC)
M680I (nt 2040 ATG->ATA)
M680L (nt 2038 ATG->CTG)
M694I (nt 2082 ATG->ATA)
K695R (nt 2084 AAG->AGG)
R761H (nt 2282 CGT->CAT)
A744S (nt 2230 GCC->TCC)
S675N (nt 2024 AGC->AAC)
∆I692 (nt 2076-2078del)
∆M694 (nt 2078-2080del)
T681I (nt 2042 ACT->ATT)
Exon 2
E148Q (nt 442 GAG->CAG)
E167D (nt 501 GAG->GAC)
T267I (nt 800 ACA->ATA)
Exon 5
F479L (nt 1437 TTC->TTG)
Exon 3
P369S (nt 1105 CCC->TCC)
Allèles complexes
E148Q-V726A
E148Q-M694V
E148Q-P369S
E148Q-∆I692
E167D-F479L
P369S-R408Q (nt 1223 CGG->CAG)
E148Q-P369S-R408Q
4. Épidémiologie
4.1. Influence de la population d’origine
Cette maladie touche essentiellement les
populations du pourtour méditerranéen, en
particulier les populations arméniennes,
turques, juives séfarades, et arabes, dans les-
quelles l’estimation de la fréquence des hété-
rozygotes pour une mutation du gène en
cause varie de 1/6 à 1/20.
Toutefois, la non appartenance à l’une de ces
populations n’est certainement pas un critère
d’exclusion du diagnostic de FMF.
4.2. Âge de début
Les premières crises apparaissent une fois sur
deux dans les dix premières années de la vie,
et dans plus de huit cas sur dix avant l’âge de
vingt ans. La maladie peut se révéler au cours
de la première année de la vie.
4.3. Sexe
La proportion observée est généralement de
l’ordre de trois hommes atteints pour deux
femmes, ce qui conduit à supposer soit une
pénétrance plus faible du phénotype chez les
femmes, soit une plus grande fréquence
d’avortements spontanés des fœtus de sexe
féminin porteurs de deux allèles mutés.
4.4. Prévalence dans la population
générale
La prévalence de la FMF dans les populations
à risques (arméniennes, turques, juives séfa-
rades et arabes) varie selon les études de
1/150 à 1/1 600.
4.5. Association à d’autres maladies
L’association à un purpura rhumatoïde a
été décrite. Une étude récente a par ailleurs
rapporté une association avec les maladies
inflammatoires chroniques de l’intestin
(maladie de Crohn et rectocolite hémorra-
gique) (10).
4.6. Environnement
Les facteurs déclenchant les crises sont mal
connus ; toutefois,le stress, les règles, ou cer-
tains facteurs alimentaires ont été incriminés.
4.7. Grossesse
Avant la découverte de l’efficacité thérapeu-
tique de la colchicine, une disparition des
crises avait été rapportée durant la grossesse.
Aucune étude n’a démontré une augmenta-
tion du risque d’anomalies chromosomiques
chez les femmes enceintes poursuivant leur
traitement par la colchicine.

5. Tests génétiques
5.1. Performance pour le diagnostic
positif
L’isolement récent du gène MEFV impliqué
dans cette maladie permet d’espérer une
meilleure prise en charge des patients et de
leur famille. Il s’agit en effet d’une des rares
maladies génétiques pour lesquelles l’analyse
moléculaire apporterait le premier critère
objectif de diagnostic, et dont les consé-
quences thérapeutiques sont essentielles.
Il faut d’emblée préciser que les résultats ont
été obtenus dans le cadre de protocoles de
recherche ; aussi, à ce jour, il n’existe pas de
consensus sur les indications et l’interpréta-
tion des analyses du gène MEFV en pratique
clinique. Les informations suivantes peuvent
cependant être rapportées :
• La valeur diagnostique de l’analyse molécu-
laire a été clairement démontrée par une
étude (4) menée dans une large population
de patients d’origine arménienne (n=90)
présentant les critères cliniques de Livneh et
appartenant à des familles dans lesquelles
aucune étude de liaison préalable (mar-
queurs en 16p13) n’avait été réalisée (iden-
tification des mutations des deux allèles
MEFV chez 89 % des patients).
•
La valeur diagnostique de ces tests moléculaires
dans d’autres populations reste à évaluer.
• Si cette valeur diagnostique est bien confir-
mée, il sera alors envisageable d’évaluer « en
retour » les critères cliniques actuels à l’aide
de tests moléculaires objectifs. Ces études
pourraient conduire à l’identification de
patients porteurs de deux allèles MEFV
mutés et présentant une symptomatologie
atypique ou fruste, non identifiable selon les
critères de Livneh. La reconnaissance d’une
telle situation aurait très certainement des
conséquences thérapeutiques essentielles. À
cet égard, deux allèles MEFV mutés ont été
identifiés chez certains patients qui avaient
un diagnostic peu probable de FMF selon les
critères de Tel Hashomer (11).
5.2. Intérêt pour le pronostic
Des études récentes ont clairement démontré (à
l’échelon de populations de patients) la sévérité
du phénotype associé au génotype M694V
homozygote, avec en particulier l’association à
une atteinte rénale, aux arthrites, et un âge de
début plus précoce (4/12) (associations validées
sur le plan statistique). Toutefois, à l’échelon
individuel, une amylose rénale a pu être docu-
mentée chez certains individus porteurs d’un
autre génotype, suggérant clairement l’existence
d’autres facteurs impliqués dans la survenue de
cette complication majeure. Cette hypothèse a
été récemment confirmée par l’identification,
dans la population de patients vivant en
Arménie, de deux facteurs génétiques modifica-
teurs du phénotype, le sexe des patients et leur
génotype au locus SAA1 (Serum Amyloid
Protein). Le risque de développer une amylose
réna
le
est en effet bien plus élevé chez les individus de
sexe masculin et chez ceux porteurs du génotype
homozygote SAA1
α
/
α
(13).

Recommandations générales (Société
Française de Génétique Humaine)
• L’examen des caractéristiques génétiques pose des
problèmes spécifiques soulignées par le Comité
Consultatif National d’Éthique Français dans ses
avis du 24 juin 1991 (n°25) et du 30 octobre 1995
(n°46).Il touche l’individu dans sa nature intime et
dans ses liens avec sa famille. Le résultat, quel qu’il
soit, peut avoir des répercussions sur la vie person-
nelle et familiale ; il peut être ressenti comme une
anormalité, voire une discrimination.
• La pratique des tests génétiques implique donc le
respect d’un certain nombre de règles qui sont résu-
mées ci-dessous.
• L’analyse des caractéristiques génétiques ne peut
pas être réalisée comme un examen de routine.
•
Avant le test, le sujet doit avoir compris la nature de
l’examen, la signification des résultats, et les consé-
quences éventuelles en terme de suivi ou de traitement.
• L’information doit être donnée par un médecin qui
a des compétences en génétique médicale. Elle doit
être directe et orale pour permettre un dialogue,
puis consignée sur un document écrit.
•
Le sujet doit avoir donné spécifiquement son
consentement écrit avant la réalisation du test. Une
fois testé, il peut refuser de connaître ses résultats et
son droit de ne pas savoir doit toujours être respecté
• L’annonce des résultats doit être faite directement
au sujet par un médecin qui, par sa compétence,
peut expliquer la signification des résultats.
• Le secret médical doit être respecté vis-à-vis des
tiers, y compris les autres membres de la famille.
Ces derniers ne doivent pas être sollicités directe-
ment par le médecin. Si un sujet refuse de faire
connaître à sa famille le risque révélé par le test
génétique qu’il a subi,le médecin est dans l’impos-
sibilité de contacter les apparentés. Il doit informer
le sujet testé de sa responsabilité et tout faire pour le
convaincre d’informer ses proches.
• Sauf cas très particuliers, les enfants mineurs ne
doivent pas être testés.
Recommandations spécifiques
Dans l’état actuel des connaissances fondées sur les
résultats de quelques protocoles de recherche, on
peut,de façon très schématique, proposer les recom-
mandations suivantes :
1. Devant un diagnostic de présomption (patient
présentant les critères cliniques de Livneh)
• Si l’analyse moléculaire révèle la présence de 2
allèles MEFV mutés, le diagnostic est confirmé.
• Si l’analyse moléculaire révèle la présence d’au plus
1 allèle MEFVmuté, le diagnostic n’est en aucun cas
infirmé.
•
Dans une famille dont le cas index porte deux allèles
mutés identifiés, si des signes cliniques évocateurs du
diagnostic de FMF apparaissent chez un membre de la
fratrie, une analyse moléculaire chez cet apparenté
concluant à la présence des deux mêmes mutations
permet très rapidement d’affirmer le diagnostic.
2.Toutefois,l’interprétation des résultats doit tenir
compte des éléments suivants
•
À ce jour, en dehors de la description récente d’un allèle
MEFV porteur d’une mutation stop en 3’ du gène
(Y688X) (14),
il n’existe pas de mutation non ambi-
guë (absence de mutation stop,frameshift, large délé-
tion...). Aussi, en l’absence de test fonctionnel, il est
extrêmement difficile de conclure à l’implication de
telle ou telle mutation dans la survenue de la maladie.
• La signification fonctionnelle des mutations identi-
fiées est d’autant plus difficile à évaluer que certains
allèles
MEFV
portent plusieurs (deux voire trois)
mutations en cis, ce qui complique également l’inter-
prétation des analyses génétiques (à ce jour, en toute
rigueur, l’identification de deux mutations chez le cas
index ne justifie pas de stopper l’analyse moléculaire :
il faut déterminer si ces mutations sont localisées ou
non sur le même allèle, et éventuellement cribler la
totalité des exons codants et des séquences d’épissage à
la recherche d’autres mutations).
• La pénétrance de cette affection est incomplète. Il
reste d’une part à évaluer ce paramètre par des
études de populations, et d’autre part à mieux pré-
ciser a posteriori le phénotype de ces individus
asymptomatiques sur le plan clinique et porteurs de
deux allèles MEFV mutés (en particulier, existence
ou non d’un syndrome inflammatoire biologique
qui, compte tenu de l’existence de formes cliniques
révélées par une amylose rénale inaugurale, pour-
rait faire discuter les indications thérapeutiques).
• Il existe une variabilité phénotypique inter et intra-
familiale.
•
Il existe des modes de transmission pseudo-dominants.
•
Enfin, devant l’absence d’autre critère objectif de
jugement, il est probablement important de rééva-
luer le diagnostic de FMF à la lumière des résultats
de l’analyse du gène
MEFV
, en tenant compte, en
particulier,des limites de cette analyse (analyse foca-
lisée sur les sites les plus fréquemment mutés,analy-
se directe «exhaustive »,ou exclusion de liaison…).
Recommandations
Auteurs Cécile Cazeneuve1, Catherine Dodé2,Marc
Delpech2,Isabelle Touitou3,Gilles Grateau4,
Serge Amselem1
1Service de biochimie génétique
Hôpital Henri Mondor – 94010 Créteil
2 Service de biochimie génétique
Hôpital Cochin – 75014 Paris
3 Laboratoire de génétique moléculaire et chromo-
somique
Hôpital A.de Villeneuve – 34295 Montpellier
4 Service de médecine interne
Hôpital de l’Hôtel-Dieu – 75001 Paris
Membres de la commission ayant participé à l’éla-
boration de la fiche
Roland Berger, Dominique Bonneau,
Françoise Clerget, François Cornelis,
Josué Feingold, Henri Plauchu, Claude Stoll,
Christine Verellen, Jacqueline Yaouanq.
Secrétariat de la commission
François Cornélis
Unité de génétique des maladies communes de l’adulte
Hôpital Lariboisière
2, rue Ambroise-Paré
75010 Paris
Document mis à jour le 5 janvier 2001
 6
6
1
/
6
100%