Maladie Périodique (ou Fièvre méditerranéenne familiale, FMF)

77
••••••••
dufaitde lagravitédelanéphro-
pathie. Sa survenueplus fréquentedans
certainesethniespourraitêtresous
l’influenceconjointedefacteurs d’en-
vironnements etde facteurs génétiques
en recherche d’identification. Introduite
en 1972,lacolchicine est toujours le
traitementde référencedelaFMF.En
priseorale continue,elle prévientla
survenuedescriseschezlamajorité
despatients ainsiqueledéveloppe-
mentde l’amylosesecondaire. Maladie
àdébut essentiellementpédiatrique,la
FMF n’étaitautrefoisévoquée quepar
élimination sur labased’arguments
cliniquesrétrospectifset/oude tests
thérapeutiques.Leretarddiagnostique
étaitlarègle etlespatients subissaient
nombred’explorationsinutiles.
L’identification dugène MEFV (pour
MediterraneanFeVer)en1997,parun
consortiumfrançais[17]etunconsor-
tiuminternational[18], apermisla
miseaupointdupremierdiagnostic
de certitude en révélantdesmutations
chez70%despatients présentantune
FMF typique. Deplus,lesétudesfonc-
tionnellesde laprotéine correspon-
dante(pyrine pour fièvre,dénommée
aussimarenostrine pour marenostrum)
ontétélepointde départ de découver-
tessur larégulation de l’inflammation
etde l’apoptose. Eneffet,cetteprotéine
Maladie Périodique
(ouFièvre
méditerranéenne
familiale,FMF)
intervientdanslavoie signalétiquedes
caspasesaboutissantentreautres,à
l’activation dufacteur de transcription
pléïotropiqueNF- κ Betàlasynthèse
d’interleukine 1,quiest une cytokine
pro-inflammatoiretrèspyrogène.
Histoirenaturelle
de lamaladie
Ledébut
La FMF est une maladie majoritaire-
mentpédiatrique. Lespremièresmani-
festationsde laFMF apparaissenten
moyenne àl’âge de 4ansetdans80%
descasavantl’âge de 20 ans.Undébut
plus exceptionnel,aucours de lapre-
mièreannée de vie ouaprèsl’âge de
50ans,doitfairediscuterune autre
causedefièvrepériodique.
Lescrises
La FMF est caractérisée pardesaccès
aigus,en apparenceauto-déclenchés,
etentrecoupésde phasesde latence.
La crisesurvientbrusquementetatteint
rapidementson acmé. Elle est souvent
Introduction
La fièvreméditerranéenne familiale
(FMF)est considérée comme le prototype
desmaladies«auto-inflammatoires».
Lenom de «auto-inflammatoire» aété
créé pour caractériserungroupe de
pathologiesliéesàundéficitde l’im-
munitéinnée paranalogie aux mala-
diesauto-immunesliéesàundéficit
de l’immunitéacquise[10].Ils’agit
d’une maladie génétiqueautosomique
récessiverépanduedanscertaines
populations,particulièrementchez
lesArméniens,lesTurcs,lesarabeset
lesjuifsd’AfriqueduNord. La première
description de laFMF est apparuedans
lalittératuremédicale dèsle début du
siècle. Lenom actuel de fièvremédi-
terranéenne familiale est plus répandu
queceluidemaladie périodique,uti-
liséplutôten France[5].
Sesmanifestationscliniquessontparo-
xystiquesetapparemmentsanscause
apparente. La fièvre,maîtresymptôme,
dureenviron 48 heuresets’accompa-
gne de douleurs abdominalesettho-
raciquestraduisantl’inflammation des
séreuses.Lescrisessontsuiviesd’une
période de latencededurée variable
pendantlaquelle le patientresteleplus
souventasymptomatique. C’est l’amy-
losesecondairequidomine le pronostic
Isabelle TOUITOU
(Montpellier),
Isabelle KONE-PAUT
(Kremlin-Bicêtre)
Tirésàpart:Isabelle Touitou,Unitémédicale desmaladiesauto-inflammatoires,Laboratoire
de génétique,HôpitalArnauddeVilleneuve,371,avenueduDoyen Giraud,34295 Montpellier
cedex5.

sonsluiayantvalule qualificatif de
«pseudo-palustre».Elle dureen
moyenne 48-72 heuresavecune
défervescenceprogressivedurant
quelquesheures.Son caractèreisolé
n’est cependantpasexceptionnel et
l’interrogatoiredoitrechercherdans
cettesituation en plus desfrissons,une
sensation de froid précédentlaphase
de sudation etuncaractèrerépétitif.
Despousséesfébrilesde basgrade,pas-
santinaperçuesoudurantseulement
quelquesheuressontaussipossibles.
Lessignesabdominaux
Ilsconstituentaveclafièvre,lessignes
cardinaux de laFMF,présents chez85
à90%desmalades.La symptomato-
logie est trèsaiguëavecune douleur
brusquelocalisée àl’épigastreetqui
diffusesecondairementàl’hypo-
chondredroit,lafosseiliaquedroite
puisàl’ensemble de l’abdomen. Le
patientaune attitude prostrée évitant
le moindrechangementde position.
Dessignesdigestifsàtype d’anorexie,
de constipation,de nauséesetplus
exceptionnellementde vomissements
sontassociés.Acestade,l’examen
abdominalmontreune distension dif-
fuseavecune sensibilitéàlapalpa-
tion quirévèle une défenseouparfois
une contracture,localisée ougénéra-
lisée. Siune radiographie d’abdomen
sanspréparation est pratiquée,elle
montreune distension digestiveavec
parfoisdesniveaux hydro-aériques
faisantredouterune occlusion intes-
tinale.
Cetableautrèsimpressionnantet
surtout lorsqu’il est inaugural,peut
appelerdesinterventionschirurgicales
(en particulierdesappendicectomies)
inutiles.Letaux d’appendicectomies
est élevéchezcespatients périodiques:
50%en1964[15], maissemble dimi-
nueravecune meilleurereconnais-
sancedelamaladie :15 %dansune
série marseillaisedel’année 2000 [7].
Quand une laparotomie est pratiquée
en crise,elle montreune congestion
péritonéale avecépanchementde
liquide trouble stérile contenantde la
fibrine etdesleucocytes.L’appendice
etle restedesorganessonten règle
normaux.Desbridesetdesadhérences
péritonéalespeuventrésulterde l’orga-
nisation de l’exsudatproduitlors de
lacriseinflammatoireetêtredans
de rarescas,àl’origine d’occlusions
intestinales.
L’examen histologiquedupéritoine en
crisemontreune inflammation asso-
ciantcongestion vasculaire,œdème et
infiltratde cellulesmononuclééeset
despolynucléaires.Enl’absenced’in-
tervention,ladouleur cède en six
àdouzeheures.Sa disparition est
complèteen24à48 heures,accom-
pagnée souventd’une diarrhée tran-
sitoire.
Lescrisesabdominalespeuventêtre
atypiquesparleur intensitéréduiteà
type de gène abdominale plutôtque
de franche douleur ouparleur locali-
sation avecpossibilitédecrisespel-
viennesévoquantune urgenceurinaire
ougynécologique.
LorsquelaFMF est connueetquela
criseabdominale est typique,le patient
peut reconnaîtresessymptômescequi
permetd’attendrelesquelquesheures
critiquesavantde prendreune déci-
sion d’intervention chirurgicale. L’état
généralconservéetlaprésenced’un
signe d’accompagnementextraabdo-
minalcomme une douleur thoracique
ouune arthritepeuventpermettrede
reconnaîtreplus aisémentlaFMF.
Danslesautressituationsoùune
urgencechirurgicale ne peut êtreéli-
minée,sediscutel’utilitéd’une ima-
gerie complémentaire. L’échographie
abdominale,examen simple etnon
irradiant,manquemalheureusement
de spécificitédanscecontexte. Pour
certains,le scannerabdominalpeut
êtreutile caril permetde vérifierla
normalitédel’appendiceetl’absence
d’occlusion intestinale. Ilpeut mon-
trerd’autressignesévocateurs d’une
crisedeFMF plutôtqued’une urgence
chirurgicale chezunsujetsain comme
unépanchementpéritonéaldefaible
abondance(70 %),desadénopathies
mésentériques(45 %) etune spléno-
mégalie (20 %) [23].
Lessignesarticulaires
Lessignesarticulairesde laFMF
incluentdesarthralgiesetdesarthrites
dontladistribution peut êtreoligo-
articulaire(80%descas)oupoly-
précédée de prodromes:irritabilité,
fatigue,sensation de malaise,endo-
lorissementabdominalvagueavec
attitude voûtée caractéristiquechezle
jeune enfant,etparfoisconstipation.
Lescrisessontvariablesen fréquence
eten durée d’unpatientàunautreet
chezunmême patient.Leterme de
fièvrerécurrenteest doncmieux adapté
que«périodique».Desfacteurs déclen-
chantsontparfoisretrouvés:émotion,
stress (peur d’unexamen parexemple),
activitéphysiqueinhabituelle,examen
para-cliniqueinvasif,menstruations
chez17%despatientes.
Entrelescrises,l’étatgénéralest conservé
maischezcertainsenfants,leur répéti-
tion est responsable d’unretardstaturo-
pondéraletde troublespsychologiques
avecanxiétéetirritabilité.
Evolution
L’évolution de laFMF est imprévisible
etdifférenteselon lespatients.Après
une phasedelatencede3à4ansen
moyenne,lescrisess’installentpen-
danttoutelavie avecune tendance
vers l’extinction définitiveavecl’âge.
Leprofil descrisespeut varierdansle
tempschezunmême patientcequi
rend difficile ladescription d’unprofil-
type. Certainspatients onttrèspeude
criseset/oudescrisestrèsatténuées
quirendentleur vie tout àfaitsup-
portable d’autantquelessignesont
tendanceàdiminueraprès40ans.
D’autres,vivantunvéritable «étatde
malpériodique»,ontdescrisessévères
quasiquotidiennesetinsensiblesaux
thérapeutiques.Danstous lescasde
figure,de longuesrémissionssontpos-
sibles.Lepronosticdépend essentiel-
lementde lasurvenued’une amylose
quisemble liée àlasévéritédela
maladie :âge de début,intensitéet
fréquencedescrises,etàl’ethnie
d’origine dupatient.
Sur quelséléments
cliniquesévoquer
laFMF?
La fièvre
La fièvretypiques’élèvebrutalement
à39-40°Cets’accompagne de fris-
78
••••••••

une spondyloarthropathie HLA B27
négativeouune colopathie inflam-
matoire. Letraitementdesarthro-
pathiesprolongéesest difficile carles
AINS etlescorticoïdesne sontpas
efficaces.Letraitementchirurgical,
synovectomie ouune miseenplacede
prothèse,peut êtreleseulrecours devant
une destruction articulaireaccélérée
et/ouune impotencefonctionnelle
absolue.
Lessignesmusculaires
Lesmyalgiesainsiquedesdouleurs
desplantesetdestalonssontprésentes
chezenviron 20 %desmalades.
Lesmyalgiesspontanéesatteignent
surtout lesmollets quisontdoulou-
reux àlapression etpeuventêtreaug-
mentésde volume,tendus etchauds.
Cesmyalgiesrégressentspontanément
en quelquesheuresoujours sans
qu’aucuntraitementne soitréellement
efficace. Lesenzymesmusculairessont
normalesoutrèspeumodifiéesavec
élévation parfoisdiscrètedelalactico-
déshydrogénase(LDH). L’imagerie et
labiopsie musculairesontnormales.
Lesmyalgiesinduitesparl’effort et
surtout parl’endurancesontlesplus
fréquentes.Ellessurviennenten général
le soir,sontd’intensitémodérée etdis-
paraissentspontanémenten quelques
heures.
Lesyndrome desmyalgiesfébrilespro-
longéessurvientpresquetoujours chez
despatients dontlaFMF est connueet
traitée parlacolchicine. Lesdouleurs
musculairessontintensesetdiffuses
confinantle patientaulit.Bien qu’ex-
ceptionnelles,lesmyalgiesfébrilespro-
longéesdoiventêtrereconnuespour
éviteraupatientdesexplorationsinutiles
etinstaurerrapidementune courtecor-
ticothérapie quiest le seultraitement
efficace.
Lessignespleuraux
Ilssontprésents chez50%despatients
souffrantde FMF,etsetraduisentpar
une douleur basithoraciqueunilatérale
(gauche le plus souvent)associée une
polypnée superficielle. Lessignesres-
piratoiresprédominentchezle très
jeune enfantquin’exprime qu’excep-
tionnellementune douleur thoracique.
articulaire(20 %). Ilsinaugurentla
maladie une foissur quatre,posant
alors chezunenfantle problème du
diagnosticdifférentiel avecune arthrite
juvénile idiopathiqueouen fonction du
contexte,avecunrhumatisme arti-
culaireaigu.
Lesformesrécurrentesmonoarti-
culairesetcellesavecatteintesimul-
tanée de deux articulationssontde
loin lesplus fréquentes,lesarticula-
tionslesplus touchéesétantlesgenoux
etleschevilles.Lesattaquesarticulaires
débutentbrutalementavecune dou-
leur atteignantson paroxysme en
moinsde 12heuresetrégressanten
moinsde 24heures.L’impotencefonc-
tionnelle etlessignesinflammatoires
locaux persistentpendantenviron
deux semaines.Cescrisesaiguës
concernentparfoislesgrossesarti-
culationsdesmembresinférieurs dont
parfoislesarticulationssacro-iliaques
etlesmétatarsophalangiennes.Elles
n’entraînenten généralpasd’érosions
osseuseset/oude destruction arti-
culaire.
Lesatteintespolyarticulairespeuvent
intéresserdesarticulationsplus ser-
réescomme lesdoigts,lespoignets et
lestemporo-mandibulaires; leur évo-
lution est plus prolongée durant6à
12semaines.Lesradiographiesprati-
quéesmontrentessentiellementun
œdème despartiesmolles,etune dis-
crètedéminéralisation osseusequin’est
quetransitoire.
Desarthropathiesprolongéessontrap-
portéesmaisellesne dépassentpas5%
de l’ensemble desatteintesarticulaires
liéesàlaFMF.Ellessiègentsurtout
aux hanchesetaux genoux (75%
descas),pouvantdurerpendantdes
semainesoudesmoismaisne laissant
généralementpasde séquellesarti-
culaires.Toutefois,desévolutionstrès
rapidesvers ladestruction articulaire
ontétérapportées,en particulierau
niveaudeshanchesaveccoxiteradio-
logique:pincementde l’interligne,
déminéralisation de voisinage pouvant
évoluervers une condensation secon-
daireetunremaniementarthrosique
desberges.Une nécroseaseptique
ischémiquedelatêtefémorale est
aussipossible. Cesmanifestationspeu
communesdoiventfairerechercher
une maladie associée,en particulier
L’irradiation desdouleurs àl’épaule ou
àlabasethoraciquecontrolatérale est
possible. L’auscultation en crisepeut
retrouverune diminution dumurmure
vésiculaire. La radiographie duthorax
montrerarementune lame d’épan-
chementpleuralet/ouune bande d’até-
lectasie persistantentredeux etdix
jours.Lescrisespleuralesaccompa-
gnentlescrisesabdominalesquipeu-
ventlesmasquer,etsuiventleur évo-
lution en 24à48 heures.
Lessignescutanés
Quand ilssontsoigneusementrecher-
chés,ilssontprésents chezunpatient
sur deux.
La plaqueérysipélatoïde est laplus
caractéristiqueparcequ’elle n’est
observée dansaucune autremaladie
auto-inflammatoire. Souventdéclen-
chée parlamarche oulastation debout
prolongée,elle siège au-dessous du
genou:région prétibiale,dorsale du
pied etpérimalléolaire. Son aspectest
érythémateux,induré,chaudetdou-
loureux.Degrande taille (15 à50cm?),
elle ne peut passerinaperçuedumalade
etconstitueunélémentanamnestique
majeur pour le diagnosticde FMF.Sa
durée est d’environ 3à4jours.
D’autrestypesd’érythèmesuniquesou
multiplescorrespondantàdesplacards
cellulo-dermiquessontdécrits au
niveaudutronc, desquatremembres,
duvisage etducou.
Despousséesd’œdèmesindolores«non
inflammatoires»,duvisage,dudosdes
mains,desmollets fontpartie inté-
grantedessignescutanésde laFMF.
Desaccèsurticariensaccompagnant
lescrisessontparfoisrapportés.
La survenuedelésionsmuqueuses
(aphtosebuccale,oubucco-génitale),
de nodulessous-cutanés(oud’éry-
thème noueux)etde purpuravasculaire
infiltréetdécliverelie cliniquementla
FMF aux vascularitessystémiques:
purpurarhumatoïde,PAN, maladie de
Behçetetcolitesinflammatoires.
Spléno-hépatomégalie
Une splénomégalie est constatée chez
25% desmaladesentrelescrises.Elle est
beaucoupplus fréquentechezl’enfant
79
••••••••

inflammatoireseten particulierde pro-
téine SAA.Sa fréquenceest variable en
fonction desethnieseten fonction du
lieude vie despatients,plus fréquente
chezlesTurcs(60 %) etlesjuifs
Sépharades(27 %) quechezles
ArméniensetlesAshkénazes.
Lemode de révélation habituel de
l’amyloseest une protéinurie non
sélectives’aggravantprogressivement
vers unsyndrome néphrotiqueetune
insuffisancerénale sur une période de
2à17ans.Alaphased’état,une
hépatomégalie etune splénomégalie
sontprésentesdansplus de 70 %des
cas.L’infiltration intestinale est très
fréquente,rarementmanifestée parun
syndrome de malabsorption. La plus
longuesurvie despatients grâceà
l’hémodialyseetàlatransplantation
faitapparaîtred’autreslocalisations
extrarénalesde l’amylosecomme des
goitresetdesvalvulopathiesamyloïdes.
Lediagnosticde l’amyloseest confirmé
dans80%descasparlabiopsie rénale
et/ourectale (moinsnécessairement
médullaire) aprèscoloration parle
rouge Congo quimeten évidencedes
dépôts biréfringents.
La dialyseetlatransplantation rénale
permettentd’allongerlasurvie des
patients périodiques.
Sur quelséléments
biologiquesévoquer
laFMF?
Comme pour laclinique,lesmodifi-
cationsbiologiquessontparoxys-
tiques; cependant,leur normalisation
complèteentrelespousséesn’est pas
larègle.
Syndrome inflammatoire
biologiquenon spécifique
Ilest caractéristiqueaumomentdes
poussées.Lesanomalieslesplus
constantestouchentcertainesprotéines
de phaseaiguë:protéine C-réactive,
fibrinogène,protéine SAAetvitesse
de sédimentation quiest supérieureà
20 mm/h. 25% despatients gardent
une protéine C-réactive,unfibrino-
gène,une VS etune protéine SAA
élevée en phaseinter-critique. Une
inflammation sub-aiguëpersistante
peut aussiêtreobservée chezdescol-
latéraux dupremierdegrédespatients,
cliniquementasymptomatiques.L’effet
de lacolchicine,trèspartiel sur les
anomaliesbiologiques,n’affecterait
quelestaux de protéine C-réactivelors
despoussées.
Sur le planhématologique
La leucocytosepeut atteindre20 000/
mm3maiselle est inconstanteet
de durée brève(<24h). Lestaux de
plaquettesetd’hémoglobine restent
normaux.
Donnéesgénétiques
Mode de transmission
La FMF est considérée comme une
maladie de transmission récessive
autosomique[13].Lerisquepour un
couple porteur d’avoirunenfantatteint
est doncde unquart.Cerisqueest à
moduleren fonction de lapénétrance
desmutations,définie comme lapro-
babilitéd’êtreaffectésionpossède une
mutation dansle casdesmaladiesà
transmission dominante,oudeux
mutationspour une maladie récessive.
Iln’est pasrarecependantde constater
unaspectde pseudo-dominance,c’est-
à-direune transmission d’aspectver-
tical. Ilfaut alors penseràrechercher
une consanguinitéouune forteendo-
gamie,union préférentielle ausein
d’une même population àrisque,ces
deux causesaugmentantconsidéra-
blementlaprobabilitédecoexistence
de deux allèlesmutéschezlesindi-
vidus issus de tellesfamilles.
Al’inverse,danslespetitesfamilles,
il n’est pasraredeneretrouveraucune
notion familiale,etlanotion de cas
sporadiquenedoitpasfaireéliminer
lapossibilitéd’une FMF.
Legène de laFMF
Legène MEFV est localisésur le
brascourt duchromosome 16,en
16p13.3[12].Detaille moyenne,envi-
ron 14 kilobases,il comprend 10exons.
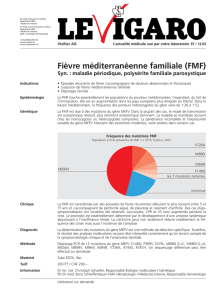
Une centaine de variants de séquences
(14-53%). Elle est pratiquement
constanteencasd’amylose.
Une hépatomégalie est détectée chez
15 à55%despatients,le plus souvent
en rapport avecune stéatosehépa-
tique,quand il n’yapasd’amylose.
Manifestationsparoxystiques
diverses
La péricarditeest une complication
bien connuedelaFMF maiselle est
relativementrareavecune prévalence
estimée à0,7%.
Signesneurologiques:méningites
aseptiquesrécurrenteschezdesmala-
descéphalalgiquesounon,ayantété
prévenuesefficacementparlacol-
chicine. Quelquesrarespatients ont
présentédescrisescomitiales
Orchite:Safréquenceest diversement
rapportée (5-20 %despatients mâles)
selon lesauteurs.Son évolution est
spontanémentrégressiveen48-
72 heures; lescomplicationsisché-
miquessontexceptionnelles.
Desmanifestationsoculairesàtype
d’épiscléritesetd’uvéitesantérieures
outotalesontétérapportées.Leur évo-
lution aétérégressivesous traitement
parcolchicine etcorticoïdes.
Complication vers l’amylose
L’amyloseest lacomplication évolutive
laplus gravedelaFMF carelle conduit
rapidementàl’insuffisancerénale ter-
minale etaudécèsdupatient.Sa sur-
venueseraitfavorisée parl’homo-
zygotie M694Vdansle gène MEFV [14]
etpardesmutationshomozygotesd’un
autregène,le gène SAA[2].Enprin-
cipe,elle survientaprèsplusieurs
annéesd’évolution (10à15 ansen
moyenne) etson incidenceest large-
menten corrélation avecl’âge des
patients puisque75% descassontdia-
gnostiquésaprès43ans.Cependant
danssaglobalité,elle atteintsurtout
dessujets jeunesde moinsde 40ans.
Elle peut exceptionnellementappa-
raîtretrèstôtdansl’enfance,avantles
manifestationscliniquesde laFMF
(amylosedetype II),maislaréalitéde
cettesituation est aujourd’huidiscutée
carpour laplupart,cessujets ontdes
taux sériquesélevésde protéines
80
••••••••

séquençage aux techniquesde détec-
tion spécifiquedemutations.Trois
troussescommercialiséesne permet-
tentd’analyserqu’unnombrelimité
de mutations.Lescinq mutationsles
plus fréquentessontsystématiquement
exploréesdanspresquetous leslabo-
ratoirescarellesrendentcompteàelles
seulesde plus de 80%descasde FMF
danslespopulationsàrisque.
La découvertededeux mutationssigne
undiagnostic«positif »,sous réserve
de confirmation quecesdeux muta-
tionsne sontpasportéesparle même
chromosome (casdesallèlescomplexes).
Cephasage peut sefaireaisémentpar
l’analysedel’ADN parental. Silesdeux
mutationssontalléliques(en cis),la
confirmation génétiquedudiagnostic
de FMF impliquel’identification d’une
troisième mutation sur l’autrechro-
mosome (en trans). La non détection de
deux mutationsparl’utilisation d’un
test génétiquelimitéàuncriblage par-
tiel dugène MEFV ne doitpasêtre
considérée comme untest «négatif ».
Danscecas,le test doitêtreinterprété
comme «non contributif »,etle dia-
gnosticde FMF ne peut êtreformelle-
mentexclu.
L’analysedugène est précieuseprin-
cipalementdanslesformespauci
symptomatiques.Beaucoupdiscutent
l’intérêtdutest génétiquedansles
populationsnon méditerranéennesen
raison dufaible taux de découvertede
mutationschezcespatients.Chezles
non atteints,certainsprônentl’ana-
lysedetous lesmembresde lafratrie
lorsquelediagnosticest confirmé
génétiquementchezle propositus.
D’autressuggèrentde testerde façon
systématiquetous lescouplesd’ori-
gine juivenon ashkénaze,arménienne,
arabeouturque,en raison dutaux par-
ticulièrementélevé(1sur 3à1sur 10)
de porteurs sainsdanscespopulations.
L’intérêtde testerlesnon atteints est
lasurveillance,notammentde lafonc-
tion rénale,avantl’apparition poten-
tielle dessymptômes.Cetteindication
dépassemalheureusementlespossibi-
litésfinancièresetorganisationnelles
de laplupart deslaboratoires.Acette
limite,serajoutenaturellementune
dimension éthique. Iln’yapasde
consensus quantàlaconduiteàtenir
dansle casde découvertefortuited’une
aétérépertoriée dansle registreinter-
netdesmutationsauto-inflammatoires
(Infevers):http://fmf.igh.cnrs.fr/infevers/
[19].Ilconvientde noterquetous les
variants dugène n’ontpasle même
effetsur le tableaucliniquedespatients.
Pour une bonne interprétation dutest
génétique,il faut différencier:
LES MUTATIONS PATHOGÈNES
L’exemple type est M694Vdans
l’exon 10,transformantlaméthionine
en position 694 en valine. Bien qu’il
n’yaiteuencoreaucune étude fonc-
tionnelle,laquasi-systématiqueasso-
ciationd’untableautypiquedeFMF
aveccettemutation lorsqu’elle est en
double dose,permetd’affirmerqu’il
s’agitd’une mutation vraie. La grande
majoritédesmutationsFMF est ponc-
tuelle,de type substitution. Acejour
n’ontétérapportéesquetroispetites
délétionsne décalantpasle cadrede
lecture,etune mutationaboutissantà
lacréation d’uncodon stop (Y688X).
LES MUTATIONS ÀFAIBLE PÉNÉTRANCE
OU POLYMORPHISMES FONCTIONNELS
Ils’agitde variants plus oumoins
associésàune symptomatologie de
FMF.Ilsontune prévalenceélevée dans
lapopulation générale. E148Qdans
l’exon 2dugène MEFV faitcouler
beaucoupd’encre. Cevariantest alter-
nativementconsidérécomme une muta-
tion vraie ouunpolymorphisme.
Despolymorphismessimples,chan-
geant(R202Q)ounon (P706P)l’acide
aminé codé,ontaussiétédécrits dans
le gène MEFV.
Lediagnosticmoléculaire
Letest génétiqueconstitueleseul
élémentbiologiquespécifiquedela
maladie périodiqueetdevraitpro-
gressivementsesubstitueràde grosses
explorationslourdesetcoûteuses,et
pourraitsefairedeplus en plus tôt
dansl’évolution de lamaladie. Il
faut cependantsoulignerquel’outil
moléculairen’apasrévolutionné le
diagnosticde laFMF autantqu’on
l’attendait.
Iln’yapasde méthode consensuelle
àcejour.Lesstratégiesmoléculaires
dépendentdeshabitudesetdusavoir-
fairedeslaboratoires,allantducri-
blage exhaustif de certainsexonspar
FMF génétiquenon exprimée clini-
quementchezunindividu:faut-il
traiterousimplementsurveiller?
Diagnosticprénataletpré-implanta-
toirenesontnaturellementpasindi-
quésdanscettepathologie,quibien
quetrèsinvalidante,n’obèrepasde
façon majeurelepronosticfonctionnel
dupatient,etquibénéficie d’untrai-
tementtrèsefficace.
Corrélationsphénotype-
génotype
Degrandesvariationspeuventexister
danslaprésentation cliniquedes
patients,non seulemententredes
maladesde différentesorigineseth-
niques,maisaussidemême génotype
MEFV,voireausein d’une même
famille. Cesdifférencespeuventêtre
en partie expliquéesparl’hétérogénéité
génétique(alléliqueetde locus),les
gènesmodificateurs,etlesfacteurs
environnementaux.
Touteslesmutationsdansle gène
MEFV n’ontpasuneffetpathogène
équivalent.La mutation M694Vest
réputée laplus sévère(âge de début
plus précoce,plus de signesarticulaires
etcutanés,risqueimportantd’amy-
loserénale). Cetteobservation aété
répétitivementfaitechezlesjuifs,les
Arméniens,etlesarabes.L’association
de M694Vàune forme plus gravede
lamaladie est controversée chezles
Turcs.V726AetE148Qontglobale-
mentuneffetmodéré.
Desanalysesde liaison génétiquedans
une population turqueontmontré
l’existencedefamillesFMF non liées
augène MEFV [1].Cetteétude initiale
n’ajamaisétésuivie de ladécouverte
dugène en causedanscesfamilles.
Alalueur desdonnéesrécentes,il est
possible quecespatients souffrent
d’une autrefièvrepériodique.
Lesgènesmodificateurs sontdesfac-
teurs génétiquesàeffetmodéréquine
sontpasindispensablespour lasur-
venuedelamaladie. Deux équipes
françaisesontmontrél’existencede
gènespouvantinfluencerl’évolution de
lamaladie indépendammentde MEFV.
MICA (MajorHistocompatibility
Complexclass Ichain A)etforme cli-
niquedelamaladie :L’effetde M694V
81
••••••••
 6
7
8
6
7
8
1
/
8
100%