Lire l`article complet

Mal prise en charge mais surtout mal
connue du corps médical, des mé-
decins experts et du personnel soi-
gnant en général, la SLA touche tout de même
8000 personnes en France soit un Français dia-
gnostiqué toutes les trois heures.
Qu’est-ce que la SLA ?
En 1865, Charcot décrivait un syndrome très
lentement évolutif, associant une atteinte pyra-
midale, avec une raideur des quatres membres,
et un syndrome pseudobulbaire, dû à une sclé-
rose isolée des faisceaux latéraux de la moelle
épinière. Quatre ans après, il décrivait la forme
classique de la SLA, associant non seulement la
sclérose des faisceaux latéraux médullaires mais
aussi une atteinte des motoneurones spinaux
entraînant une atrophie musculaire diffuse et
rapidement évolutive. Cette dernière fut consi-
dérée par Erb comme une nouvelle entité cli-
nique et nommée sclérose latérale primitive
(SLP), maladie dix fois plus rare que la SLA.
La SLA se caractérise donc par l’atteinte des mo-
toneurones, cellules nerveuses qui commandent
et contrôlent les muscles de la motricité volon-
taire. Cette maladie est évolutive et entraîne la
paralysie des membres, mais aussi celle des
muscles de la phonation et de la déglutition. Les
malades, qui conservent leurs facultés intellec-
tuelles, sont emmurés dans leur corps, et l’attein-
te des muscles respiratoires est la dernière phase
avant le décès. Cette maladie cruelle désoriente
l’entourage et le personnel soignant, dont les in-
firmières libérales quelquefois confrontées à ce
problème qui est le plus souvent traité à domi-
cile. L’association SLA souhaite la généralisation
d’un poste d’infirmier référent, déjà présent au
groupe hospitalier de la Pitié-Salpêtrière à Paris.
Car le premier handicap est la formation insuffi-
sante des acteurs paramédicaux mais aussi des
médecins. En effet, le malade consulte au début
pour un trouble considéré par lui-même comme
bénin, d’origine ORL, pneumologique, rhumato-
logique... Quelques signes vont alerter le méde-
cin. Ainsi, la SLA touche les adultes entre 50 et
70 ans, mais des cas chez les personnes plus
jeunes sont de plus en plus dénombrés. La mala-
die peut débuter par une paralysie d’un segment
de membre ou par un trouble de la parole ou de
la déglutition. La forme spinale (premier cas) est
plus communément observée chez les hommes et
conduit progressivement à une incapacité d’usage
des membres inférieurs et supérieurs. La forme
bulbaire touche plus volontiers les femmes, et les
premiers symptômes sont une difficulté à parler
ou à avaler, une hypersialorrhée (augmentation
de la salive). Au début de la maladie, les patients
peuvent ressentir des crampes et/ou des fascicu-
lations qui sont des contractions de faible ampli-
tude de fibres musculaires isolées, bien diffé-
rentes par exemple des myokymies. Un autre
symptôme peut être associé : la sensation d’en-
raidissement articulaire définissant la spasticité
qui s’accentue dès que le patient veut entamer un
mouvement. Une vivacité des réflexes tendineux
ou, à l’inverse, leur diminution peut être obser-
vée. On observe aussi une constipation, des
troubles du sommeil d’origines diverses, une hy-
persialorrhée, des douleurs surtout articulaires
et également une fragilité émotionnelle trop vive.
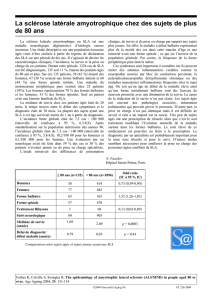
En bref, le diagnostic, à la fois simple et com-
pliqué, s’établit sur les atteintes des motoneu-
rones corticaux, spinaux ou bulbaires. Des at-
teintes cognitives, des atteintes des sphincters
ou du système sensitif, visuel ou auditif ne sont
pas des symptômes de la SLA. Un électromyo-
gramme (EMG) est nécessaire pour confirmer le
diagnostic. De nouvelles méthodes d’imagerie
comme l’IRM, la spectro-IRM et la stimulation
magnétique peuvent également être utiles.
Lucie Galion
Information SLA : www.ars.asso.fr
Un colloque a été organisé à Paris par l’Association
pour la recherche sur la sclérose latérale amyotro-
phique (SLA). Un livre noir a été édité, mettant en évi-
dence les dysfonctionnements des organismes sociaux
et médicaux dans la prise en charge de cette maladie.
37
Maladie orpheline
La SLA tient ses états généraux
1
/
1
100%