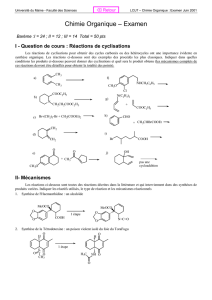
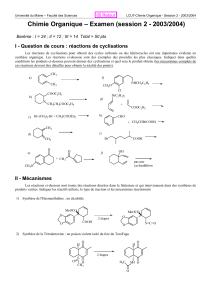
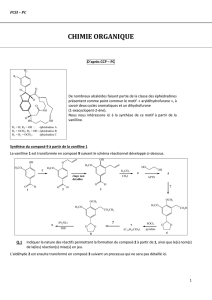
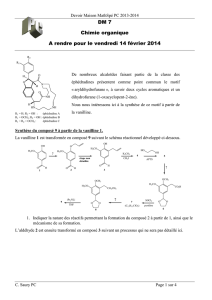
Correction du devoir surveillé n°6 - PCSI Option PC

PC A - PC B CHIMIE - DS n°6 - Correction
1
Partie 1 :
Préparation d’un tampon phosphate
D’après l’épreuve spécifique PC du concours ENSTIM 2008
1. Si l’on veut une quantité totale en phosphore égale à c, il faut introduire :
m
1
=
cVM
1
=
0
,
10
×
1
×
336
=
33
,
6
g
.
2. Compte-tenu des différents pK
a
de l’acide, on peut tracer le diagramme de
prédominance de cet acide. On trouve donc que les espèces présentes au pH considéré
(6,2) sont HPO
42-
et H
2
PO
4-
.
pH
pKa
pKa pKa
12 3
H
3
PO
4
H
2
PO
4
HPO
4
2
PO
4
3
H
3
PO
4
=
H
2
PO
4
=
H
2
PO
4
HPO
4
2
=
PO
4
3
HPO
4
2
2.1 7.2 12.4
3. Lorsqu’on ajoute l’hydroxyde de sodium la réaction est :
H
2
PO
4-
+ OH
-
= HPO
42-
+ H
2
O. Sa constante est :
K=Ka2
Ke=10−7,2
10−14 =106,8 >>1.
On peut donc la considérer comme totale. (Et l’espèce en défaut est OH
-
.)
4.
On déduit du tableau d’avancement de la réaction précédente :
On en déduit :
[HPO
42−
]=
n
2
V
et
[H
2
PO
4−
]=
n
1
−
n
2
V
.
(Le mélange qu’on obtient suite à cette réaction totale est un mélange de deux espèces
conjuguées qui coexistent. Le RP suivante : H
2
PO
4-
+ HPO
42-
= HPO
42-
+ H
2
PO
4-
ne
modifie pas la composition.)
5.
Alors le pH de la solution s’exprime :
2
2
1 2
n
pH pKa log
n n
= +
−
.
6.
On trouve :
n
2
n
1
−n
2
=10
pH−pK
a2
=10
6,2−7,2
=0,1=
1
10
.
On en tire :
n
2
=
n
1
11
=
0
,
1
11
=9,0.10
-3
mol
.
Alors
m
2
=n
2
M
2
=
0
,
1
11
×40 =0,36 g
.
Samedi 28 mars 2008
DS n°6
Chimie Organique et Chimie des Solutions aqueuses
Durée : 2 heures
CORRECTION

PC A - PC B CHIMIE - DS n°6 - Correction
2
Partie 2 :
Synthèse de l’éthoheptazine
D’après Centrale
1)
L’ion amidure est une base forte de pKa
≈
35. Il se produit une réaction acido-basique
conduisant à un carbanion
B
fortement stabilisé par mésomérie :
NNNNN
2) C
=
B
r
N
= 2-bromo-N, N-diméthyléthylamine.
3) B
est un bon nucléophile qui va réaliser une S
N
2 sur
C
. (On privilégie aussi le
mécanisme S
N
2 ici car l’halogénoalcane est primaire.)
N
Br N
N
N
Br
-
D
D
est traité à nouveau par de l’amidure de sodium pour donner un anion analogue à
B
. Puis on
ajoute du 1-bromo-3-chloropropane et on isole alors un produit
E
(C
15
H
21
N
2
Cl).
4)
On forme à nouveau un carbanion sur le carbone voisin du cycle et on procède à une
seconde S
N
2 :
N
N
Br
-
E
Br Cl
N
N
Cl
5)
On procède à une seconde S
N
2 sur le carbone électrophile porteur du brome car il est
meilleur nucléofuge que le chlore (la liaison C-Br est plus polarisable que C-Cl).

PC A - PC B CHIMIE - DS n°6 - Correction
3
E
est chauffé pendant 16 heures à 100°C pour conduire à un intermédiaire
F
(C
15
H
21
N
2+
, Cl
-
) ; puis le mélange est chauffé à 225°C pendant 2 heures, ce qui libère
du chlorométhane et permet d’isoler
G
(C
14
H
18
N
2
).
6)
Formules développées de
F
et de
G
:
Cl
-
F
N
N
G
N
N
7)
Ecrire les mécanismes réactionnels des passages
E
→
F
et
F
→
G
.
E
N
N
Cl
Cl
F
N
N
G
N
N
S
N
2 intramoléculaire S
N
2
Cl
8)
On doit chauffer car ces deux réactions sont difficiles. En effet, la formation d’un
grand cycle (7 chaînons) n’est pas très facile et l’ion chlorure n’est pas un bon
nucléophile.
G
est chauffé pendant 3 heures à 120 °C en présence d’une solution d’acide sulfurique
concentré pour conduire à
H
(C
14
H
19
NO
2
).
9)
On réalise ici l’hydrolyse de la fonction nitrile de
G
en acide carboxylique :
H
N
O
OH
Le mélange précédent est à nouveau chauffé pendant 16 heures en présence d’un excès
d’éthanol, ce qui conduit à l’éthoheptazine recherchée.
10)
On réalise une
estérification
avec l’éthanol :
Ethohéptazine
N
O
O

PC A - PC B CHIMIE - DS n°6 - Correction
4
Partie 3 :
Synthèse du calacorène
1.
Formules topologiques des composés D, E, F, I, J, K
1
et K
2
:
B
r
M
g
D
M
g
B
r
E
F
O
H
I
N
B
r
M
g
J
O
K
1
O
C
l
O
Cl
K
2
Majoritaire Minoritaire
2.
Schéma réactionnel de la réaction E
→
F :
MgBr
OOMgBr
O
M
g
B
r
Mg
2+
OH
H
+
Br
-
3.
J possède
4
stéréoisomères (car un carbone asymétrique + une double liaison C=C
stéréogène).

PC A - PC B CHIMIE - DS n°6 - Correction
5
R-E
O
S-E
O
R-Z
O
S-Z
O
Enantiomères
Diastéréoisomères
4.
Mécanisme de la réaction J
→
K
1
: il s’agit d’une addition électrophile ionique sur un
alcène dont la régiosélectivité est dictée par la loi de Markovnikov généralisée : le
produit majoritaire est issu de l’intermédiaire réactionnel le plus stable.
K
1
O Cl
O
Cl
K
2
Majoritaire
Minoritaire
J
OH Cl
O
Cl
O
et
Cl
Minoritaire
car plus proche du groupe C=O,
qui exerce un fort effet inductif électroattracteur,
donc déstabilise l'espèce électrodéficitaire qu'est le carbocation
Majoritaire
 6
7
8
9
10
11
12
13
14
6
7
8
9
10
11
12
13
14
1
/
14
100%