Télécharger - Thèses d`exercice de Santé

1
UNIVERSITE TOULOUSE III PAUL SABATIER
FACULTE DES SCIENCES PHARMACEUTIQUES
ANNEE : 2014 THESES 2014 / TOU3 / 2079
THESE
POUR LE DIPLOME D'ETAT DE DOCTEUR EN PHARMACIE
Présentée et soutenue publiquement
par
GREGOIRE HELENE
COMPARAISON DE DEUX TYPES DE CELLULES SOUCHES PLURIPOTENTES :
EMBRYONNAIRES ET INDUITES.
Date de soutenance
Directeur de thèse : Couderc Bettina
JURY
Président : Couderc Bettina
1er assesseur : Taboulet, Florence
2ème assesseur : Parini Angelo
3ème assesseur : Petel Alain

2
REMERCIEMENTS
Au terme de ce travail, je tiens à remercier l’ensemble des personnes qui ont contribué
à ce projet. Je remercie tout particulièrement les membres de mon jury de thèse d’avoir
accepté d’examiner cette thèse.
Un grand merci à ma directrice de thèse le Pr. Bettina Couderc de m’avoir fait
connaître ces cellules au cours d’une soirée au Café Bioéthique Etudiant. Merci de m’avoir
accompagnée et dirigée tout au long de ce travail qui m’a passionné.
Je remercie également le Pr. Angelo Parini, le Pr. Florence Taboulet et M. Alain Petel
d’avoir accepté de faire partie de mon jury. Un grand merci au Pr. Florence Taboulet d’avoir
été là tout au long de ces six années d’études, de m’avoir permis d’apporter une petite aide au
Café Bioéthique Etudiant et de m’avoir fait confiance pour exposer ses réflexions lors d’une
conférence d’Arboris Scientae. Merci à Alain Petel pour les promenades dominicales à
Balma, merci aussi de m’avoir permis de travailler dans votre pharmacie pour mon tout
premier contrat en tant que pharmacien.
Je remercie énormément mes parents et mes sœurs, pour leur patience et leur amour
depuis toujours. Merci de m’avoir soutenu tout au long de ces études, et merci à mes parents
et à Estelle d’avoir pris le temps de corriger avec justesse ma thèse. Merci à mes grands-
parents pour l’amour qu’ils me donnent sans compter. Merci à Marie qui veille si bien sur
moi.
Merci à mes amies de pharmacie : Marie, Julie, Marine, Amélie, Charlotte, Anne,
Faustine et Laurène pour votre joie, votre présence et tous ces moments partagés que je
n’oublierai jamais. Merci à Célia, Bastien, Najib et Pol-André, les soirées salsa partagées avec
vous me manqueront.
Merci à mes compagnes de révisions Raphaëlle, Clarisse et Fanny. C’est bien grâce à
vous et à votre soutien que je suis arrivée là ! L’amitié que vous m’offrez est ce que j’ai de
plus cher. Merci à Emilie pour ton écoute et ta compréhension, Marie-Elisabeth pour ta

3
solidarité et ton soutien, Marie-Eléonore pour toutes ces années, Samuel F. pour ta gentillesse.
Merci à Yaëlle pour les fous rires, Mathilde et Philippe pour notre amitié d’Eyne à Toulouse
en passant par Strasbourg, Samuel M. pour les riches conversations que nous avons eu et ton
indéfectible amitié. Un grand merci à FX pour ton incomparable prévenance et ta présence et
à Marie-Cécile de m’avoir donné ta si précieuse amitié depuis plus de vingt ans.
Merci à mes chères voisines Paula, Maya et Mme Amiel, j’étais vraiment chez moi ici.
Aujourd’hui je pars, mais je ne vous oublierai pas à l’autre bout du monde !

4
« Sapience n’entre point en ame malivole,
et science sans conscience n’est que ruyne de l’ame. »
Rabelais (Pantragruel, 1532)

5
Table des matières
TABLE DES MATIERES ....................................................................................................................................... 5
TABLE DES ILLUSTRATIONS ............................................................................................................................... 7
LISTE DES ABREVIATIONS ................................................................................................................................. 9
INTRODUCTION .............................................................................................................................................. 11
PREMIER CHAPITRE : CELLULES SOUCHES ET PLURIPOTENCE ...................................................................... 12
A. LES CELLULES SOUCHES ................................................................................................................................... 12
1) Cellules souches : définitions ............................................................................................................... 12
a) Les deux caractéristiques principales de la cellule souche ............................................................................... 12
b) Démontrer qu’une cellule est pluripotente ..................................................................................................... 14
i. Chez la souris ............................................................................................................................................... 14
ii. Chez l’homme .............................................................................................................................................. 15
2) Les différents types de cellules souches ............................................................................................... 16
a) Les différents degrés de potence ..................................................................................................................... 16
b) Les cinq types de cellules pluripotentes ........................................................................................................... 17
i. Les cellules embryonnaires de carcinome (ECC) ......................................................................................... 18
ii. Les cellules souches embryonnaires (ESC) .................................................................................................. 18
iii. Les cellules souches de l’épiblaste (EpiSC) .................................................................................................. 19
iv. Les cellules embryonnaires germinales (EGC) ............................................................................................. 19
v. Les cellules pluripotentes induites (iPSC) .................................................................................................... 19
B. LES CELLULES SOUCHES PLURIPOTENTES ............................................................................................................. 20
1) Induction expérimentale de la pluripotence ........................................................................................ 20
a) La fusion cellulaire ............................................................................................................................................ 21
b) Le transfert nucléaire somatique (SCNT) ......................................................................................................... 22
c) La reprogrammation induite ............................................................................................................................ 23
d) La transdifférenciation intratissulaire .............................................................................................................. 24
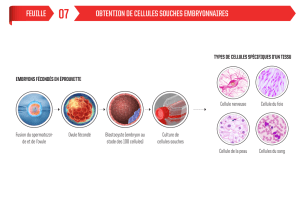
2) Les cellules souches embryonnaires ES ................................................................................................ 24
a) Origine des blastocystes ................................................................................................................................... 25
b) Isolement des cellules souches de l’ICM .......................................................................................................... 26
c) Mise en culture des cellules ES humaines. ....................................................................................................... 27
d) Vérification de leur transformation effective ................................................................................................... 28
3) Les cellules souches pluripotentes induites iPS .................................................................................... 29
a) Choix des cellules reprogrammées................................................................................................................... 29
b) Choix des facteurs de reprogrammation .......................................................................................................... 30
c) Choix de la méthode de vectorisation .............................................................................................................. 33
i. Méthodes avec intégration ......................................................................................................................... 33
ii. Méthodes sans intégration.......................................................................................................................... 34
iii. Méthodes sans vecteurs .............................................................................................................................. 35
DEUXIEME CHAPITRE : COMPARAISONS ENTRE CELLULES ES ET CELLULES IPS ............................................ 38
A. UTILISATION DES CELLULES SOUCHES ................................................................................................................. 38
1) Les cellules souches pluripotentes : un outil précieux pour la recherche ............................................. 38
a) La modélisation pathologique .......................................................................................................................... 39
b) Le screening ..................................................................................................................................................... 40
c) En toxicologie ................................................................................................................................................... 41
2) Les cellules souches pluripotentes, un outil en thérapie cellulaire et médecine régénératrice. .......... 41
a) Définitions ........................................................................................................................................................ 41
i. La thérapie cellulaire ................................................................................................................................... 42
ii. La médecine régénératrice .......................................................................................................................... 43
b) Les domaines d’application en thérapie cellulaire ........................................................................................... 44
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
1
/
118
100%