Le glucose - anarlf.eu

Le cerveau et le glucose….
F. Lenfant
Le pronostic des traumatismes crâniens graves ou des accidents vasculaires cérébraux est lié à
l’importance de la lésion primaire mais également à la survenue d’évènements susceptibles de
favoriser le développement de lésions secondaires évoluant vers l’ischémie. Classiquement, ces
évènements regroupent différents facteurs comme l’hypotension artérielle, l’hypoxie, l’hypo- ou
l’hypercapnie, l’hyponatrémie et l’hypo- ou l’hyperglycémie. Au cours de lésions cérébrales, les
perturbations de la glycémie sont fréquentes et, parfois, la conséquence directe de l’atteinte
neurologique. Récemment, le contrôle strict de la glycémie chez les patients de réanimation a été
remis à l’ordre du jour1. Une telle stratégie a permis, en effet, d’améliorer considérablement le
pronostic de ces patients2. Toutefois, le problème posé par le contrôle de la glycémie chez les
patients présentant une atteinte cérébrale reste complexe.
Le présent exposé fera, dans un premier temps un rappel physiologique sur le glucose, sa
régulation et son métabolisme cérébral, et abordera dans un deuxième temps les conséquences
des perturbations de la glycémie sur le cerveau lésé et, enfin, tentera de dégager une attitude
pratique pour les patients.
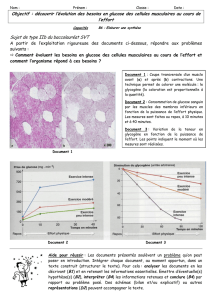
Régulation de la glycémie
La glycémie est régulée principalement par l’insuline qui est secrétée en abondance de manière à
faire pénétrer le glucose dans les organes insulino-sensibles. En période post-prandiale, lors du
pic glycémique, l’insuline est secrétée en abondance. Associée à l’hyperglycémie, elle va

favoriser l’oxydation du glucose et va permettre d’orienter l’excès de substrats énergétiques vers
le stockage en inhibant la libération des AG du tissu adipeux, en stimulant la synthèse du
glycogène, en accélèrant le transport du glucose dans le muscle, en favorisant la synthèse des
triglycérides et la captation des triglycérides par le tissu adipeux, et en inhibant la
néoglycogénèse, et la glycogénolyse.
De nombreux facteurs peuvent concourir à la survenue d’une hyperglycémie chez le sujet
agressé. La glycémie dépend avant tout des apports exogènes en glucose, de la production
endogène hépatique et/ou rénale, de l’insulinémie et de la non-réponse des organes à l’insuline
constituant l’insulino-résistance. Lors d’une agression, il semble qu’apparaissent précocement
des perturbations du métabolisme glucidique. La principale cause d’hyperglycémie chez le
patient agressé est l’insulino-résistance et l’augmentation de la production endogène de glucose.
De nombreux facteurs semblent impliqués, mais ce de façon plus ou moins prouvée. Il s’agit de
la stimulation sympathique, de la sécrétion hormonale en réponse au stress (cortisol, glucagon),
mais également des cytokines.
La glycolyse
D’une façon générale, le glucose est un nutriment pour toutes les cellules. Son oxydation, en
bicarbonate ou en lactate, permet la production d’ATP, nécessaire au bon fonctionnement de la
cellule et des enzymes consommatrices d’énergie. Cette voie métabolique suit une succession de
réactions biochimiques, la glycolyse cytoplasmique, le cycle de Krebs, et la chaîne respiratoire
métabolique. Schématiquement, le glucose traverse la membrane cytoplasmique grâce à des
protéines transporteuses et est transformé en glucose-6-phosphate (G6P). Ensuite, le G6P va être
métabolisé, et conduit à la formation de pyruvate. En présence d’oxygène, le pyruvate va entrer
dans la mitochondrie et être métabolisé en acétate qui conduira à la formation d’acétylcoA qui

sera secondairement métabolisé dans le cycle de Krebs. Le bilan de la glycolyse aérobique est la
somme des bilans de chaque étape. Une mole de glucose permet la formation de 38 moles d’ATP.
En absence d’oxygène, les lactates déshydrogénases forment la dernière étape de la glycolyse et
la formation de lactate en est la résultante. En ce cas, le bilan énergétique est pauvre, 2 moles
d’ATP pour une mole de glucose.
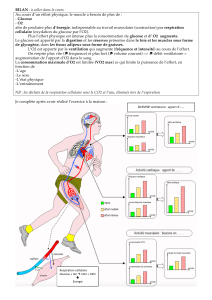
Cerveau et particularités métaboliques
Si le cerveau est un grand consommateur d’énergie (20 à 25% de la production quotidienne
d’ATP), il est pratiquement dépourvu de forme de stockage d’énergie (glucose, glycogène et
oxygène). En revanche, les neurones peuvent utiliser le lactate, le pyruvate et la glutamine (mais
non les acides gras) comme source d’énergie3,4. L’insuline, quant à elle, n’a pas d’effet sur le
métabolisme énergétique cérébral5. Par ailleurs, le cerveau se caractérise par la présence de la
barrière hémato-encéphalique et de plusieurs contingents de cellules différents. La paroi des
artérioles et des capillaires cérébraux est tapissée d’un endothélium non fenêtré, formant la
barrière hémato-encéphalique dont la caractéristique fonctionnelle principale est sa perméabilité
sélective. Le passage d’une substance du plasma sanguin vers le parenchyme cérébral dépend de
son coefficient de liposolubilité. Le glucose ne peut franchir cette barrière que s’il est véhiculé
par des transporteurs membranaires spécifiques. Les jonctions serrées entre les astrocytes et le
fait qu'ils constituent un syncytium contribuent à isoler l'espace extracellulaire périneuronal de
l'espace péri-vasculaire. Par conséquent, le glucose sanguin est transporté et métabolisé
essentiellement par ces cellules.
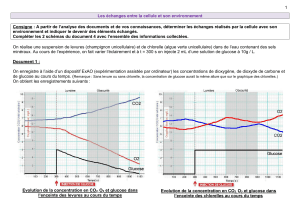
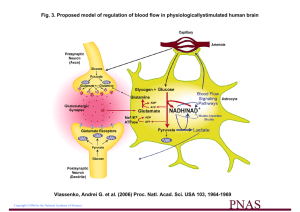
Les astrocytes transforment le glucose par la voie de la glycolyse. Le flux de la glycolyse dans
ces cellules étant très élevé, une grande partie du glucose est finalement transformée en acide
lactique grâce à la lactate déshydrogénase (LDH). Le lactate, une fois relâché dans l'espace

extracellulaire périneuronal par les astrocytes, est immédiatement repris par les neurones qui
possèdent les transporteurs spécifiques nécessaires6. Dans les neurones, le lactate est transformé
en pyruvate par la LDH. Le pyruvate entre alors dans le cycle de Krebs mitochondrial qui
alimente la chaîne respiratoire produisant l'ATP. Ainsi, le lactate fourni par les astrocytes à partir
du glucose semble être le substrat métabolique principal des neurones. L’abondance de lactate et
donc de pyruvate permet de produire, en plus de l’énergie, des acides aminés, comme le
glutamate, qui est le principal neurotransmetteur excitateur dans SNC et le précurseur immédiat
de la formation du GABA, un neurotransmetteur inhibiteur. Ceci n’est possible dans les neurones
que si le cycle de Krebs tourne en plein régime. Dans les astrocytes, où il y a transformation
massive du pyruvate en lactate, le glutamate entre dans le cycle de Krebs comme un
intermédiaire de remplacement. Le glutamate relâché dans la fente synaptique par les neurones
est recapté par les astrocytes qui possèdent au niveau de leur membrane plasmique, surtout dans
la partie située à proximité des synapses, de puissants transporteurs de glutamate. Le glutamate
est alors transformé en glutamine dans les astrocytes par l'enzyme glutamine synthétase (GS). La
glutamine, quant à elle, est facilement relâchée dans l'espace extracellulaire. La majeure partie de
la glutamine est ensuite captée par les neurones où elle contribue à la formation du
neurotransmetteur glutamate. Il est possible que le glutamate transporté dans l'astrocyte lors d'une
intense activité synaptique neuronale provoque une activation de la glycolyse et donc de la
production d'acide lactique, substrat utilisé par les neurones. Ainsi, l'apport de substrat
métabolique aux neurones serait régulé par le signal « glutamate », que les neurones enverraient
aux astrocytes.
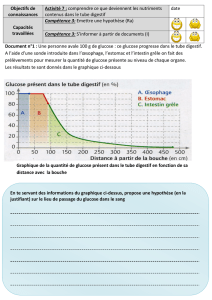
En situation d’ischémie, la glycolyse conduit à la production de lactate qui ne peut participer à la
production d’énergie. Il en résulte une acidose cellulaire et une forte production de glutamate. La
composante excitotoxique liée au glutamate augmente alors de façon dramatique. En effet, le

glutamate dépolarise le neurone et déclenche des potentiels d’action. Les récepteurs NMDA
activés par la dépolarisation laissent entre le Ca2+. Le neurone fait donc face à une charge
métabolique importante pour maintenir ses équilibres ioniques, alors qu’il ne peut produire
qu’une faible quantité d’énergie. Cela conduit alors à la mort cellulaire.
En situation d’hypoxie, la glycolyse anaérobie serait à même de produire l’énergie nécessaire à la
survie cellulaire. Toutefois, cela ne tient pas compte de l’accumulation de lactate ni de la
surproduction de glutamate.
Glucose et lésion cérébrale
Le traumatisme crânien grave est responsable d’importantes modifications neuro-hormonales
ayant un impact sur la régulation de la glycémie et le métabolisme du glucose. Ces modifications
consistent en une élévation précoce du taux d’adrénaline et de noradrénaline circulantes7 et une
augmentation de la sécrétion de cortisol, de glucagon ainsi que d’insuline. Néanmoins ces
modifications ne semblent pas spécifiques au traumatisme crânien, car également observées lors
de traumatismes graves sans lésion cérébrale8. Ceci se traduit par des perturbations du
métabolisme avec, une diminution de la production hépatique de glucose et de la lipolyse
associée à une hyperglycémie et une hypoglutaminémie traduisant une altération du relargage de
la glutamine par le cerveau9. Lors d’un traumatisme crânien, on observe également une sécrétion
de cytokines propres à modifier le métabolisme glucidique et entraîner une insulino-résistance.
Ces cytokines sont également à l’origine d’une altération de la barrière hémato-encéphalique et
de modifications du métabolisme cellulaire10,11. Ces faits expliquent donc la forte incidence de
l’hyperglycémie lors des traumatismes crâniens grave.
Parallèlement, la survenue de lésions cérébrales est susceptible d’entraîner de profondes
perturbations du métabolisme énergétique cérébral. Lors d’un traumatisme crânien expérimental,
 6
7
8
9
10
11
12
13
14
15
6
7
8
9
10
11
12
13
14
15
1
/
15
100%