le syndrome de Guillain-Barré, diagnostic et traitements

Revue
mt 2011 ; 17 (1) : 3-10
Polyradiculonévrite aiguë :
le syndrome
de Guillain-Barré,
diagnostic et traitements
David Orlikowski , Tarek Sharshar
Service de réanimation, hôpital Raymond Poincaré, 104 boulevard Raymond Poincaré,
92380 Garches, France
<david.orliko[email protected]>
Le syndrome de Guillain-Barré (SGB) est la première cause de paralysie aiguë extensive dans
les pays industrialisés depuis la disparition de la poliomyélite. Son diagnostic reste essen-
tiellement clinique. C’est une maladie évolutive et un examen neurologique précis et répété
permettra le plus souvent de confirmer le diagnostic, de juger de l’évolutivité de la maladie
et donc de sa gravité potentielle. Les examens complémentaires sont essentiellement utiles
pour éliminer une autre pathologie, notamment une méningoradiculite par la ponction lom-
baire ou une atteinte médullaire par une IRM. Les éléments essentiels de la prise en charge
sont initialement symptomatiques, par l’identification des patients à risque de développer une
insuffisance respiratoire aiguë et secondairement par la prévention et la prise en charge des
complications liées aux paralysies. L’autre volet thérapeutique est le choix entre échanges
plasmatiques et les immunoglobulines intraveineuses.
Mots clés : paralysie ascendante, polyradiculonévrite, insuffisance respiratoire aiguë,
échanges plasmatiques, immunoglobulines intraveineuses
Depuis l’éradication de la polio-
myélite antérieure aiguë, le
syndrome de Guillain-Barré (SGB)
est devenu la cause la plus fré-
quente de paralysie aiguë et extensive
dans les pays industrialisés. La gra-
vité de ce syndrome est variable
et une ventilation mécanique est
nécessaire dans 30 % des cas [1-
3]. On estime que 5 % des patients
risquent de décéder de cette maladie
[4, 5] et que 10 à 20 % garderont
des séquelles définitives [6]. C’est
une urgence neurologique dont le
diagnostic reste essentiellement cli-
nique. La prise en charge consiste
d’abord à en établir le diagnos-
tic, d’autant que diverses pathologies
du système nerveux périphérique ou
central peuvent donner un tableau
clinique similaire et que le tableau
initial du SGB peut être incomplet
voire atypique. La seconde étape va
être d’évaluer la sévérité et le risque
évolutif potentiel notamment déter-
miner si le patient doit être admis
en réanimation, le principal risque
évolutif immédiat étant l’insuffisance
respiratoire aiguë dont la probabi-
lité de survenue peut-être maintenant
appréciée [7]. Elle se conclura par le
choix du traitement spécifique entre
échanges plasmatiques (EP) et immu-
noglobulines intraveineuses.
Épidémiologie
du syndrome
de Guillain-Barré
Le SGB est une maladie rare,
dont l’incidence dans les pays indus-
trialisés est estimée à environ 0,4-
4/100 000 habitants par an [8, 9]. Le
SGB touche tous les âges et toutes
les ethnies. En Europe, cette inci-
dence est plutôt estimée entre 1,3 et
1,9/100 000 avec une relative stabi-
lité dans le temps [10]. Sa répartition
est ubiquitaire dans le monde et est
doi:10.1684/met.2011.0309
mt
Tirés à part : D. Orlikowski
3
Pour citer cet article : Orlikowski D, Sharshar T. Polyradiculonévrite aiguë : le syndrome de Guillain-Barré, diagnostic et traitements. mt 2011 ; 17 (1) : 3-10
doi:10.1684/met.2011.0309
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

Revue
habituellement sporadique bien que de petites épidémies
secondaires à des gastro-entérites liées à la contamina-
tion de l’eau par Campylobacter Jejuni aient été décrites
en Chine [11]. Depuis une vingtaine d’années des formes
d’expression clinique et électrophysiologiques différentes
ont été rapportées [12]. Ces formes sont en particulier liées
à des agents initiaux comme le C. Jejuni ou le cytomégalo-
virus [13]. Leur incidence et leur répartition géographique
est variable dans le monde (en particulier formes axonales
en Chine). Leur pronostic est également probablement dif-
férent avec un risque fonctionnel plus important à long
terme pour les SGB associés à C. Jejuni [12].
La maladie peut être observée dès la période intra-
utérine et la période néonatale jusqu’au grand âge mais
semble prédominer à l’âge adulte et sa fréquence aug-
menter avec l’âge [14]. Curieusement, pour une maladie
de mécanisme inflammatoire, il existe une légère prépon-
dérance masculine [12]. Différentes études épidémiolo-
giques ont montré qu’un premier pic d’incidence survenait
à la fin de l’adolescence ou chez l’adulte jeune et qu’un
second pic plus tardivement vers la cinquantaine [8].
Physiopathologie
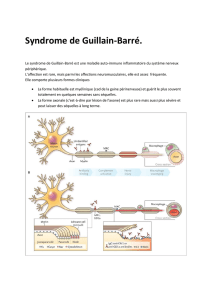
L’atteinte nerveuse est due à une infiltration par des
macrophages et des lymphocytes activés du tissu ner-
veux périphérique, s’y associe à des degrés plus ou moins
important des dépôts d’anticorps associés à une activa-
tion du complément [15]. Ces mécanismes inflammatoires
sont médiés par le biais de cytokines. Les étapes initiales
conduisant à ces réactions sont mal connues. Dans les
formes démyélinisantes il existe une infiltration multifo-
cale de cellules mononucléées. Les macrophages activés
par les lymphocytes T sont guidés par des antigènes situés
sur la myéline. Un dépôt d’anticorps et une activation du
complément entraînant une dissolution de la myéline sur-
viennent probablement à la phase initiale [16]. Les lésions
axonales sont en général secondaires à la destruction de
la myéline.
Dans les formes axonales pures (motrices ou sensiti-
vomotrices), les anticorps dirigés contre les gangliosides
nerveux se fixent directement au niveau des nœuds de
Ranvier. Il en résulte l’invasion par les macrophages acti-
vés et la destruction directe de l’axone [17].
Le mécanisme proposé est celui d’un mimétisme
moléculaire (molecular mimicry) entre des constituants
antigéniques d’un agent (le plus souvent infectieux) pro-
dromique et les constituants nerveux [18]. Il existe en effet
dans le mois précédent le SGB un événement infectieux
dans à 50 à 75 % des cas (le plus souvent respiratoire, grip-
pal ou digestif) [12] et des étiologies infectieuses à la fois
bactériennes et virales ont été incriminées comme étant
des causes certaines de SGB [18]. Bien que ces étiologies
soient reconnues, il n’existe pas de preuve qu’un traite-
ment a visée anti-infectieuse puisse améliorer le pronostic
du SGB.
Si les choses sont claires pour les formes associées à
C. Jejuni ou le mimétisme moléculaire entre le lipopo-
lysaccharide de la paroi bactérienne et les gangliosides
constituant du nerf périphérique ainsi qu’une production
d’anticorps dirigé contre ces gangliosides [19] sont prou-
vés, les choses le sont beaucoup moins pour les autres
étiologies ou l’existence d’un mimétisme moléculaire et le
rôle des anticorps antigangliosides sont beaucoup moins
nets [20]. Ainsi, des données récentes suggéreraient que
la persistance du génome du CMV dans le sang ou le LCR
serait associé à des SGB [21].
Si le CMV et le C. Jejuni représentent environ
40 % des causes de SGB, certaines étiologies comme
l’EBV et Mycoplasma pneumoniae ne représentent que
quelques pour-cent et aucune cause n’est identifiée dans
plus de 50 % des cas. Il existe néanmoins des arguments
pour penser qu’un ou des agents infectieux (de type viraux)
à tropisme respiratoire ou responsables de syndrome grip-
paux soient à l’origine de la majorité de ces cas [22].
Diagnostic
Présentation clinique
La présentation clinique la plus classique du SGB
est celle d’une paralysie extensive d’évolution aiguë (en
moins de 4 semaines) et ascendante avec un déficit glo-
balement symétrique et à prédominance proximale, une
atteinte des paires crâniennes (paralysie faciale et ou
troubles de la déglutition), des troubles sensitifs modérés
(le plus souvent subjectifs) et une abolition des réflexes
ostéotendineux. Ce tableau clinique est quasi pathogno-
monique d’un SGB quand il est complet. Du fait même
de son caractère progressif, ces éléments cliniques diag-
nostiques du SGB ne sont pas d’emblée présents, ce
qui impose donc de répéter l’examen neurologique, ce
d’autant que des formes motrices pures (le plus souvent
axonale), sensitives pures, touchant d’emblée les paires
crâniennes (avec une évolution descendante), voire ne
présentant qu’une dysautonomie isolée ont été rapportées
[10]. La réitération de l’examen clinique et neurologique
permettra également d’évaluer la rapidité d’extension du
SGB et de détecter la survenue d’une atteinte respiratoire.
Il pourra mettre en évidence des signes atypiques pouvant
faire douter du diagnostic. La répartition des déficits peut
être d’une topographie différente de l’atteinte proximale.
L’extension des paralysies peut être de vitesse (forme sur-
aiguë en moins de 24 heures ou subaiguë/chronique en
plus de 4 semaines) ou de sens différent (forme d’évolution
descendante voire bulbaire pure).
4mt, vol. 17, n◦1, jan-fév-mars 2011
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

Le diagnostic de SGB est clinique mais il ne pourra
souvent être établi de fac¸on formelle qu’au terme
d’une évolution caractéristique en trois phases, d’abord
d’extension, puis de plateau et enfin de récupération.
Une phase prodromique correspondant le plus souvent
à une infection bactérienne ou virale dans le mois pré-
cédent l’apparition des premiers signes neurologiques est
fréquente, cette phase n’est pas nécessaire au diagnos-
tic mais est évocatrice de SGB. Il est important pour le
clinicien d’avoir des critères de présomption diagnos-
tique suffisants. La classification d’Asbury et Cornblath
[23] répond à cette attente puisqu’elle regroupe, selon
le degré de plausibilité, différents paramètres cliniques,
biologiques et électrophysiologiques (tableau 1). Elle pro-
pose également des critères d’élimination du diagnostic.
Les examens complémentaires comme la ponction lom-
baire sont surtout indispensables pour écarter une autre
étiologie, plus particulièrement une méningoradiculite.
Évolution
La faiblesse musculaire est constatée lors de la pre-
mière consultation dans 70 pour cent des cas. Elle
intéresse initialement la partie proximale des membres
et suit une extension ascendante chez la moitié des
patients. Les formes motrices pures sont décrites dans
18 pour cent des cas et seraient préférentiellement asso-
ciées à une infection par Campylobacter jejuni. Cinquante
à 80 % des patients se plaignent de paresthésies qui
respectivement dans 60 et 20 % des cas précèdent ou
accompagnent les signes moteurs. Elles s’étendent généra-
lement de manière ascendante. Les anomalies sensitives
objectives intéressent essentiellement la proprioception.
Une abolition des réflexes ostéotendineux dans le terri-
toire parétique est la règle. Une aréflexie généralisée est
trouvée dans 60 à 80 % des cas. Il existe très exception-
nellement un signe de Babinski. Ce signe s’il est présent
doit faire éliminer une lésion centrale (médullaire par
exemple). L’atteinte des nerfs crâniens est plus rarement
inaugurale, une extension descendante est observée dans
5 % des cas. Il s’agit essentiellement d’une paralysie
faciale, uni ou bilatérale. Les troubles de la déglutition
et de l’oculomotricité sont initialement exceptionnels [24]
mais une atteinte bulbaire est retrouvée dans environ 30 %
des cas dans notre expérience [25].
L’association d’une aréflexie, de troubles proprio-
ceptifs et une ophtalmoparésie constituent la triade du
syndrome de Miller-Fisher. Des formes se présentant ini-
tialement comme un syndrome de Miller-Fisher sont
possibles et peuvent évoluer vers un SGB. Des formes
sensitives pures de SGB, et plus exceptionnellement dys-
autonomiques pures, ont été décrites.
Des manifestations dysautonomiques peuvent être
observées essentiellement durant la phase d’ascension
des paralysies. Depuis l’introduction des traitements
spécifiques à visée immunomodulatrice, les manifes-
tations cardiovasculaires graves à type de bradycardie
extrême voire d’arrêt cardiaque sont devenues excep-
tionnelles. Les manifestations les plus fréquentes sont en
fait les troubles sphinctériens à type de rétention d’urine.
Il faut signaler que ces manifestations ne sont pas pré-
sentes d’emblée mais apparaissent secondairement en
cours d’évolution. L’existence d’une rétention d’urine très
précoce doit faire évoquer là aussi une atteinte centrale
d’autant plus que les réflexes ostéotendineux sont pré-
sents. De même il n’existe pas d’incontinence urinaire ou
anale qui doivent faire évoquer une atteinte de la queue
de cheval.
Ponction lombaire et électroneuromyogramme
L’objectif de la ponction lombaire est de recher-
cher une dissociation albumino-cytologique évocatrice
du diagnostic de polyradiculoneuropathie aiguë, bien
que non spécifique. L’absence d’hyperprotéinorachie iso-
lée n’élimine pas un SGB, car elle est retardée de 3 à
15 jours par rapport au début clinique. Une réaction
méningée modérée (<10 éléments/mm3) est possible
(tableau 1) par contre une réaction cellulaire importante
(>50 éléments/mm3) doit faire évoquer le diagnostic de
méningoradiculite. Une cytorachie entre 10 et 50/mm3
étant compatible avec un SGB, la ponction lombaire devra
être refaite 24 à 48 heures plus tard. Une diminution du
nombre d’éléments est en faveur d’un SGB.
Les anomalies éléctrophysiologiques précoces sont
une augmentation des ondes F et un bloc de conduc-
tion motrice de siège proximal et distal. L’altération de
la conduction distale et des potentiels sensitifs appa-
raît secondairement. Une dénervation est observée dans
10 % des cas. L’électroneuromyogramme permet de clas-
ser le SGB en polyradiculoneuropathie inflammatoire
aiguë démyélinisante (AIDP ; acute inflammatory demye-
linating polyradiculoneuropathy), en neuropathie aiguë
axonale motrice (AMAN ; acute motor axonal neuropa-
thy), en neuropathie aiguë axonale motrice et sensitive
(AMSAN ; acute motor and sensory axonal neuropathy)
et en forme inexcitable. Bien que le traitement spécifique
de ces entités électrophysiologiques soit identique, leur
distinction revêt une pertinence physiopathologique mais
également pronostique. En effet, la survenue d’une insuffi-
sance respiratoire aiguë est plus fréquente dans les formes
démyélinisantes [26] et les séquelles plus sévères dans les
formes inexcitables et, dans une moindre mesure, axo-
nales [27, 28].
Diagnostics différentiels
La problématique diagnostique du syndrome de
Guillain Barré est celle d’une paralysie aiguë extensive
mt, vol. 17, n◦1, jan-fév-mars 2011 5
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

Revue
Tableau 1. Critères diagnostic de Guillain Barré d’après Asbury [23].
Signes
Nécessaires au
diagnostic
En faveur Douteux Élimination
Déficit moteur
progressif de
plus d’un
membre
Clinique typique Clinique variante LCR EMG Persistance
d’une asymétrie
de l’atteinte
motrice
Exposition aux
sovants
(hexacarbones)
Progression <4
semaines
Fièvre initiale En faveur Blocs de
conduction
(80 % des cas)
Persistance de
troubles
sphinctériens
Porphyrie aiguë
intermittente
Aréflexie Relative
symétrie de
l’atteinte
Signes sensitifs
marqués
Hyperprotéino-
rachie survenant
après une
semaine
d’évolution
Diminution des
vitesses
conduction
(60 % des cas)
Troubles
sphinctériens
initiaux
Infection
diphtérique
récente
Signes sensitifs
modérés
Progression
>4 semaines
Moins de 10
éléments/mm3
Augmentation
des latences
distales
Hypercellularité
>50/mm3
Intoxication au
plomb
Atteinte des
paires
crâniennes
Absence de
récupération
(séquelles
importantes)
Variante Absence
d’onde F
Eléments
polynuclées
dans le LCR
Atteinte
uniquement
sensitive
Récupération Troubles
sphinctériens
Absence
d’hyperprotéino-
rachie
Normal
(20 % des cas)
Niveau sensitif Autres cause de
paralysie aiguë
(polyomyélite,
botulisme,etc.)
Dysautonomie
cardiovasculaire
Signes d’atteinte
du système
nerveux central
Cellurarité ente
11 et 50
éléments/mm3
Absence de
fièvre initiale
et donc de ne pas passer à côté d’une autre lésion ner-
veuse dont la prise en charge et le pronostic peuvent être
radicalement différents.
L’examen neurologique est fondamental et doit évi-
demment rechercher tout symptôme ou signe évocateurs
d’une atteinte neurologique centrale, médullaire ou encé-
phalique. L’urgence majeure est l’élimination d’une lésion
médullaire en cas de niveau sensitif, de troubles sphinc-
tériens (rétention d’urine d’emblée ou incontinence) ou
de paraparésie flasque (lésion de la queue de che-
val) s’aggravant sans que des signes neurologiques aux
membres supérieurs ou des signes céphaliques appa-
raissent. Si une lésion médullaire est suspectée, elle doit
être formellement écartée par une IRM de qualité de
l’ensemble de la moelle et relue par un neuroradiologue
expert. Un syndrome de Miller-Fisher atypique doit faire
demander une imagerie cérébrale pour rechercher une
lésion du tronc cérébral.
Devant une atteinte motrice pure, la kaliémie et les
CPK doivent être systématiquement dosées une paralysie
dyskalièmique ou une myopathie inflammatoires (poly-
myosite) sont possibles. Une anomalie de la jonction
neuromusculaire aiguë soit présynaptique (botulisme) ou
post-synaptique (syndrome myasthénique) est également
d’autres diagnostics différentiels. Toutefois, les données
anamnestiques et cliniques ainsi qu’électrophysiologiques
permettent d’en établir rapidement le diagnostic.
Devant un déficit sensitif pur, une neuronopathie sen-
sitive doit être électrophysiologiquement recherchée.
Un dosage urinaire des porphyrines peut être utile.
En effet, les porphyries sont un diagnostic difficile mais
essentiel à évoquer car le pronostic vital est en jeu et
des mesures thérapeutiques spécifiques existent. Le défi-
cit moteur s’accompagne habituellement de douleurs,
notamment abdominales, de troubles psychiatriques, sen-
sitifs, dysautonomiques et sphinctériens. L’existence d’un
6mt, vol. 17, n◦1, jan-fév-mars 2011
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

facteur déclenchant et d’urines pourpres sont des argu-
ments supplémentaires.
D’autres causes de neuropathie sont possibles notam-
ment lorsque l’examen clinique montre une atteinte de
type polynévritique avec une atteinte motrice distale
et ou un déficit sensitif franc en gant et en chaus-
sette. Une neuropathie carentielle ou toxique doivent
alors être évoquées. Certaines mono-multineuropathies
peuvent, lorsqu’elles sont très étendues et confluentes,
mimer également un SGB. Il est classiquement observé
dans les vascularites une distribution tronculaire des
anomalies neurologiques et un syndrome inflammatoire
associé ou non à une hyperéosinophilie. Cependant les
anomalies biologiques peuvent être absentes dans les
vascularites purement nerveuses. Dans ces cas (poly-
et mono/multineuropathie), la ponction lombaire est
alors normale mais l’électroneuromyogramme permet de
redresser le diagnostic. Si une vascularite est suspectée,
une biopsie neuromusculaire permettra le diagnostic et
de débuter le traitement.
Le diagnostic de SGB peut parfois s’avérer difficile
chez un patient diabétique mais en général une pro-
téinorachie élevée (supérieure à 1 g/L) est retrouvée
et l’association à des anomalies électrophysiologiques
compatibles avec une polyradiculoneuropathie aiguë
signeront le diagnostic.
Une méningoradiculite peut mimer un SGB, elle peut
être d’origine soit infectieuse (maladie de Lyme, Herpesvi-
ridae, HSV, VZV, etc.), soit inflammatoire ou néoplasique
(maladie systémique, lymphome...). Un traitement anti-
biotique et antiviral pourra être instauré jusqu’à obtention
des résultats de l’enquête diagnostique bactériologique
sérologique et les PCR
Le SGB est par définition idiopathique, ce qui implique
qu’une affection générale notamment une maladie de sys-
tème, un cancer, un lymphome ou une infection par le
virus HIV ait été récusée. Un examen général complété
par un bilan immunologique simplifié (immunoélectro-
phorèse et dosages des facteurs antinucléaires, du latex
Waaler-Rose, des fractions C3 et C4 du complément,
des complexes immuns circulants, de la protéinurie des
24 heures), hématologique (NFS, vitesse de sédimentation)
et une sérologie VIH est en pratique suffisant.
Par contre les explorations sérologiques, les recherches
virologiques et les coprocultures à la recherche d’un agent
infectieux prodromique n’ont qu’un intérêt descriptif et
éventuellement pronostique. Il en est de même pour les
anticorps anti-gangliosides bien que ceux-ci soient plus
souvent associés notamment aux formes axonales de SGB.
Il semblerait ainsi que les SGB associés à une infection
par le Campylobacter Jejuni soient par exemple plus sou-
vent moteurs purs et axonaux et qu’ils aient un pronostic
fonctionnel plus sévère [27]. À ce jour, l’identification de
l’agent infectieux n’a aucune conséquence thérapeutique.
Évaluation de la gravité
L’insuffisance respiratoire aiguë est la complication
majeure du SGB et l’identification des patients à risque
de développer une insuffisance respiratoire aiguë et donc
l’orientation vers la structure la mieux adaptée (salle de
neurologie ou de médecine, soins intensifs ou réanima-
tion) sont les points clés de la prise en charge initiale.
Après identification et orientation des patients les plus
à risques, seront démarrés les traitements symptomatiques
et préventifs (nutrition entérale, ventilation mécanique,
etc.) puis étiologiques (immunoglobulines intraveineuses,
échanges plasmatiques)
Le recours à la ventilation mécanique, nécessaire dans
20 à 30 % des cas, accroît significativement sa mor-
bimortalité. Ainsi le taux de mortalité augmente-t-il de
5 pour cent chez l’ensemble des patients SGB à 20 pour
cent chez ceux mécaniquement ventilés. La parésie des
muscles inspiratoires et expiratoires, en association avec
les troubles de la déglutition, favorise l’encombrement, la
formation d’atélectasies et la survenue d’une pneumopa-
thie d’inhalation ; la ventilation mécanique invasive celle
de pneumopathie nosocomiale [5, 25, 29].
Les facteurs de risque précoces de survenue d’une
insuffisance respiratoire aiguë ont été bien identifiés et
peuvent être recueillis y compris aux urgences :
–Délai entre le début du déficit moteur et
l’hospitalisation inférieure à sept jours :
•impossibilité à relever la tête du plan du lit ;
•capacité vitale (CV) à l’admission inférieure à 60 %
de la valeur théorique [7].
La probabilité d’être ultérieurement ventilé est de 85 %
chez les patients ayant ces trois signes [7].
En l’absence de mesure de la CV, 3 facteurs de risques
peuvent être associés.
–Existence d’une cytolyse hépatique :
•impossibilité de tenir debout [7] ;
•impossibilité de soulever les coudes [7].
Dans ce cas la probabilité de nécessité de ventilation
est également de 85 % si 4 critères cliniques sans la mesure
de la CV sont présents à l’admission.
En pratique, les patients s’aggravant vite, ayant perdu la
marche, ayant une atteinte proximale ou axiale importante
doivent être surveillés en/ou à proximité d’un service de
réanimation.
L’électroneuromyogramme est également utile. Nous
avons ainsi montré que l’insuffisance respiratoire est plus
fréquente et sévère dans les formes démyélinisantes [26].
Une fois le patient avec SGB orienté, se posera le pro-
blème de l’indication de la ventilation mécanique et du
moment opportun pour introduire celle-ci. La difficulté
d’évaluer cliniquement l’atteinte respiratoire va nécessiter
de recourir à ces tests fonctionnels comme la mesure de
la capacité vitale. En effet, l’épuisement respiratoire dans
mt, vol. 17, n◦1, jan-fév-mars 2011 7
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.
 6
7
8
6
7
8
1
/
8
100%