L*exploration du métabolisme des acides aminés

L’exploration du métabolisme
des acides aminés
4 eme année pharmacie
Dr Morsli.M

Plan
Introduction
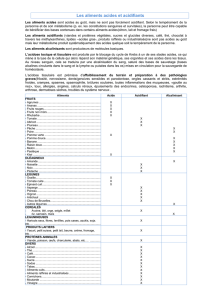
I. Structure des acides amines
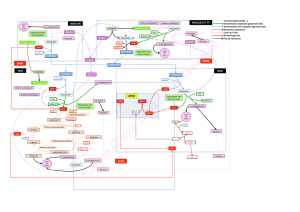
II. Métabolisme des acides aminés
III. Les pathologies du métabolisme des acides
amines
IV. Exploration des pathologies du métabolisme
des acides amines

Introduction
•Les acides amines constituent avec les glucides et les lipides
les briques constitutives de la cellule
•Ils proviennent essentiellement de la dégradation des
protéines alimentaires ainsi que des protéines endogènes ou
bien d’une synthèse de novo
acides amines
Protéines
endogènes Protéines
alimentaires
Synthèse
de novo

Introduction
La structure des acides amines leur procure des propriétés
physico-chimique qui seront utiles pour leurs mise en évidence
pour le diagnostic des aminoacidopathies, qui sont des
pathologies dues à des déficits enzymatiques des voies du
métabolisme des acides amines

structure des acides amines
•Les acides amines sont des molécules qui possèdent :
une fonction acide carboxylique
Une fonction amine primaire
Une chaine latérale R COOH
Cette structure leur procure des propriétés physico-chimiques qui sont
exploitées pour leur mise en évidence, ils existent des réactions
communes à tous les AA qui sont dues au groupement carboxyle et
amine, et des réactions spécifiques qui sont dues à la chaine
latérale R
CH
RNH3
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
1
/
48
100%