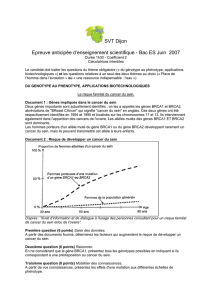
50 ans

Prédispositions génétiques aux
cancers
Dominique Stoppa-Lyonnet

T poumon, 70
T col utérus, 40 ?
T sein, 65
T foie, 80
T côlon, 65
T ORL, 70
65 ans T sein, 55
T sein, 75
? ? ?


Rétinoblastome : une tumeur rare de la rétine chez le
jeune enfant

Tumeur rare de l ’enfant : environ un enfant sur 15 à 20 000,
soit ~ 40 à 50 nouveaux cas /an
Dans 10% des cas, histoire familiale
•transmission dominante (un des deux parents atteint dans
l’enfance)
•le plus souvent atteinte bilatérale
•maladie précoce : dg avant 12 mois
Dans 90% des cas, pas d’histoire familiale
. 60% cas unilatéraux, 30% cas bilatéraux
Rétinoblastome : une tumeur rare de la rétine chez le
jeune enfant
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
1
/
35
100%