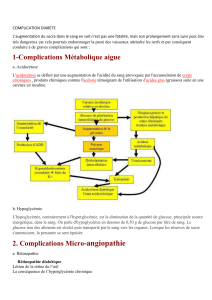
syndrome d`hyperglycemie hyperosmolaire

UNIVERSITESIDIMOHAMMEDBENABDELLAH
FACULTEDEMEDECINEETDEPHARMACIE
FES
AnnéeThèseN°/201115811
UNIVERSITESIDIMOHAMMED
BENABDELLAH
FES
SYNDROMED'HYPERGLYCEMIE
HYPEROSMOLAIRE
THESE
PRESENTEEETSOUTENUEPUBLIQUEMENTLE30/12/2011
PAR
Néele28Juillet1982àkhénifra
Mlle.ZADDOUQHANANE
POURL'OBTENTIONDUDOCTORATENMEDECINE
MOTS-CLES:
Hyperglycémiehyperosmolaire-Diabètesucré-Acidocétosediabétique-Déshydratation
JURY
M. Professeur
M. Professeur
.
Professeur
M.MADANINAOUFEL
Professeur
M.
KANJAANABIL.............................................................
d’Anesthésieréanimation
CHRAIBIABDELMJID.....................................................
d’Endocrinologiediabétologieetnutrition
MmeBONOWAFAA.........................................................
deMédecineinterne
........................................................
agrégédeRéanimationmédicale
meAJDIFARIDA...........................................................
Professeuragrégéd’Endocrinologieetmaladiesmétaboliques
JUGES
PRESIDENT
RAPPORTEUR

1
TABLE DES MATIERES
INTRODUCTION .................................................................................................. 12
PREMIER CHAPITRE : CONSIDERATIONS THEORIQUES .............................................. 23
I. RAPPEL .................................................................................................................. 24
II. DEFINITION .......................................................................................................... 26
III. ETIOPATHOGENIE ................................................................................................. 29
A- Physiopathologie .......................................................................................... 29
1- Insulinocarence ........................................................................................ 29
2- Insulinorésistance ..................................................................................... 29
a- Voie de signalisation de l'insuline .......................................................... 30
b -Rôle du tissue adipeux dans l'insulinorésistance .................................... 30
3- Rôle du rein .............................................................................................. 31
B- Hyperglycémie .............................................................................................. 31
C- Hyperosmolarité ........................................................................................... 34
1-Hyperosmolarité plasmatique ..................................................................... 34
a-Régulation de la balance hydrique ......................................................... 34
a-1-Arginine vasopressine ..................................................................... 34
a-2 La soif ............................................................................................. 34
a-3-Actions cellulaires de l'arginine vasopressine ................................... 36
a-4-Bases moléculaires de la régulation osmotique au niveau du rein ..... 36
b-Mécanismes de l'hyperosmolarité .......................................................... 37
b-1-Déficit hydrique pur ........................................................................ 37
b-2-Déficit hydrique associé à un déficit sodé ........................................ 37
b-3-Surcharge en solutés osmotiques efficaces ...................................... 37
2- Hyperosmolarité dans le SHH .................................................................... 38

2
D- Une cétogenèse inhibée ou au moins limitée................................................. 40
1-Modification de la lipolyse et de la libération des AGL ................................ 40
2-Contrôle intra hépatique ........................................................................... 41
3-Rôle de l’hyperglycémie effet anti cétogène du glucose ............................ 42
4- Rôle de la déshydratation et de l’hyperosmolarité ..................................... 43
5-Absence d’acidocétose .............................................................................. 44
E-Polyurie osmotique responsable d’une perte hypotonique d’eau et
de sodium ......................................................................................................... 47
F-Adaptation du système nerveux central à l’hyperosmolalité plasmatique ........ 47
IV. DIAGNOSTIC POSITIF DU SHH ............................................................................... 50
A- Circonstances de découverte ........................................................................ 50
B- Signes cliniques ............................................................................................ 51
C- Tableau biologique ....................................................................................... 52
1-Hyperosmolarité ........................................................................................ 52
2-Hyperglycémie ........................................................................................... 52
3-Equilibre acido-basique ............................................................................ 53
4-Déshydratation ......................................................................................... 53
5-Pool sodé ................................................................................................... 54
6-Pool potassique ......................................................................................... 55
7-Insuffisance rénale ..................................................................................... 55
8-Autres paramètres ..................................................................................... 55
9-Comparaison des paramètres biologiques du SHH et DA ............................. 55
V-COMPLICATIONS .................................................................................................. 56
A- Complications thromboemboliques ........................................................... 56
B- Hémorragies cérébrales ............................................................................... 57
C- Œdème cérébral ........................................................................................... 57

3
D- Rhabdomyolyse ........................................................................................... 58
E- Complications infectieuses ............................................................................ 58
F- Lipémie diabétique ...................................................................................... 58
G- Obstruction canaliculaire .............................................................................. 59
H- Autres complications .................................................................................... 59
VI-TRAITEMENT ....................................................................................................... 60
A-Traitement curatif.......................................................................................... 60
1-Restauration de l'équilibre hydro sodé ....................................................... 60
a-Hyperosmolarité plasmatiques sévères ................................................... 60
b-Cas du SHH. ........................................................................................... 61
2-Equilibre acido-basique ............................................................................. 63
3-Restauration des électrolytes ...................................................................... 64
a-Natrémie ................................................................................................ 64
b-Pool potassique ...................................................................................... 64
c-Pool phosphoré ...................................................................................... 66
d-Chlorémie .............................................................................................. 67
e-Magnésémie .......................................................................................... 67
4-L’Insulinothérapie ...................................................................................... 68
5-Mesures thérapeutiques générales ............................................................. 72
6-Surveillance clinique et biologique ............................................................. 72
B-Traitement préventif .................................................................................... 73
DEUXIEME CHAPITRE : PARTIE PRATIQUE
MALADES ET METHODES ...................................................................................... 74
RESULTATS ....................................................................................................... 82
I-ETUDE DESCRIPTIVE DES PATIENTS ADMIS POUR SYNDROME D’HYPERGLYCEMIE
HYPEROSMOLAIRE ................................................................................................. 83

4
A- Données épidémiologies ............................................................................... 83
1-Caractéristiques démographiques .............................................................. 83
a-Age ........................................................................................................ 83
b-Sexe....................................................................................................... 84
2-Motif d’hospitalisation ............................................................................... 85
3-Service d’admission initiale ........................................................................ 85
4-Profil évolutif du diabète ............................................................................ 86
a-Répartition selon le statut antérieur connu diabétique/non connu
diabétique .............................................................................................. 86
b-Répartition selon le type du diabète ........................................................ 86
c-Ancienneté du diabète ............................................................................ 87
d-Traitement antidiabétique antérieur ........................................................ 87
e-Suivi et complication métabolique antérieure .......................................... 88
f-Complications dégénératives associées ................................................... 88
5-Autres antécédents .................................................................................... 88
B- Données cliniques ........................................................................................ 89
1-Signes fonctionnels .................................................................................... 89
2-Signes physiques ....................................................................................... 90
a-Etat neurologique ................................................................................... 91
a-1-Score GCS ....................................................................................... 91
a-2- Signes déficitaires .......................................................................... 90
b-Score APACHEII ...................................................................................... 90
c-Etat d’hydratation ................................................................................... 91
d-Fréquence respiratoire............................................................................ 91
e-Fréquence cardiaque .............................................................................. 91
f-Tension artérielle .................................................................................... 92
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
1
/
199
100%