Comment agit la prednisone? Pourquoi pas toujours?

Forum Med Suisse 2011 ;11(27):473–477 473
curriculum
Comment agit la prednisone? Pourquoi pas toujours?
Andreas W. Jehle
Transplantationsimmunologie und Nephrologie, Universitätsspital Basel
Introduction
La prednisone est l’un des principaux représentants
des glucocorticoïdes utilisés à visée thérapeutique. Elle
agit surtout en se liant au récepteur des glucocorti
coïdes (RG) dans le cytoplasme des cellules [1]. Le RG
ainsi activé peut pénétrer dans le noyau et y réguler la
transcription de gènes. Plusieurs autres effets non gé
nomiques des glucocorticoïdes sont en outre connus qui
ne passent pas par la régulation de la transcription. Le
ligand naturel le plus important du RG est le cortisol,
synthétisé par la corticosurrénale à partir du choles
térol.
L’ effet biologique du cortisol est extrêmement poly
valent. Les glucocorticoïdes sont utilisés en pratique
courante surtout pour le traitement de nombreux pa
tients souffrant de pathologies inflammatoires et/ou
autoimmunes telles qu’asthme, arthrite rhumatoïde ou
maladies inflammatoires intestinales chroniques, où ils
sont très efficaces. D’autres maladies inflammatoires,
comme la bronchopneumopathie chronique obstructive
(BPCO), sont en majeure partie résistantes au glucocor
ticoïdes [2, 3]. Une autre limitation est celle de leurs
très nombreux effets indésirables [4]. Comme leur nom
l’indique, ils ont un effet marqué sur le métabolisme du
glucose. L’ un de leurs effets indésirables les plus fré
quents est la manifestation d’un diabète.
Après une brève introduction sur les bases molécu
laires d’une inflammation chronique, cet article présen
tera un aperçu des mécanismes d’action moléculaires
des glucocorticoïdes dans les malades inflammatoires
chroniques. Le but est d’en montrer quelques sites et
mécanismes d’action généraux, de rafraîchir les connais
sances de l’effet de cette importante classe de médi
caments et de les compléter par quelques exemples. Un
autre paragraphe expliquera le problème cliniquement
significatif de la résistance aux glucocorticoïdes à
l’exemple de la BPCO. Et enfin, une perspective sur de
tout nouveaux modulateurs sélectifs du RG et leur fort
potentiel d’améliorer le profil des effets et effets indési
rables des glucocorticoïdes.
Mécanismes moléculaires
d’une inflammation chronique
Les pathologies inflammatoires chroniques comme
l’asthme, l’arthrite rhumatoïde, les maladies inflamma
toires intestinales chroniques ou la BPCO sont toutes
caractérisées par une infiltration et une activation de
plusieurs cellules inflammatoires, qui expriment toute
une gamme de protéines inflammatoires et en sécrètent
certaines. Le spectre va des cytokines proinflamma
toires avec leurs récepteurs à l’expression de protéases
contribuant à une inflammation exagérée par la disso
lution de protéines allant jusqu’à la destruction tissu
laire, en passant par des molécules d’adhésion, impor
tantes pour l’infiltration et le homing des cellules
inflammatoires [5]. L’ expression plus marquée de ces
protéines inflammatoires est réglée au niveau de la
transcription par des facteurs de transcription proin
flammatoires. L’ un de ces plus importants facteurs est
le facteur nucléaire kappa B (NFkB), activé par exemple
dans les voies respiratoires des asthmatiques, mais
aussi dans les articulations des patients souffrant
d’arthrite rhumatoïde. L’ organisation de la chromatine
joue un rôle central dans la régulation de l’expression
de gènes proinflammatoires [6]. Les stimuli activant la
transcription de ces gènes modifient la structure de la
chromatine, processus pouvant être inversé par les glu
cocorticoïdes. Pour mieux comprendre ce processus et
l’effet des glucocorticoïdes, le paragraphe cidessous
est consacré à la structure de la chromatine.
Vous trouverez les questions à choix multiple concernant cet article
à la page 466 ou sur Internet sous www.smf-cme.ch.
Quintessence
PLes glucocorticoïdes tels que la prednisone jouent un rôle clé dans le
traitement de nombreux patients souffrant de pathologies inflammatoires
et/ou autoimmunes, mais uniquement à court ou moyen terme. Un trai
tement prolongé par glucocorticoïdes systémiques est problématique en
raison de leurs effets indésirables non négligeables. Un autre problème
est l’échec du traitement, ou résistance aux glucocorticoïdes.
PLes glucocorticoïdes sont des ligands du récepteur des glucocorti
coïdes (RG), dont il existe plusieurs isoformes. Le RG comporte en outre
des sousunités fonctionnelles modifiées de différentes manières par des
ligands naturels ou pharmacologiques. Ce qui explique pourquoi il y a un
effet biologique différent en fonction du type de cellule et du ligand.
PL’ effet classique des glucocorticoïdes est génomique, c.àd. par régu
lation de la transcription qui se fait au niveau moléculaire selon différents
mécanismes. De nombreux effets indésirables métaboliques ne se mani
festent que si le RG se lie directement à l’ADN en tant qu’homodimère.
PDe tout nouveaux modulateurs sélectifs du RG, qui n’en provoquent
pas la dimérisation, ont un bon effet antiinflammatoire sans graves ef
fets indésirables métaboliques. Le potentiel de ces nouvelles substances
est grand même si la preuve d’un bénéfice doit encore être fournie par
des études cliniques.
Andreas W. Jehle
L’ auteur n’a déclaré
aucune obligation
financière ni
personnelle en
rapport avec cet
article.

Forum Med Suisse 2011 ;11(27):473–477 474
curriculum
Régulation de l’expression génique
par la structure de la chromatine
La chromatine est faite d’ADN et d’histones (fig. 1 x).
Les histones sont des protéines formant la colonne ver
tébrale des chromosomes. Nous savons depuis long
temps qu’elles jouent un rôle critique dans la régulation
de l’expression des gènes. Dans une cellule au repos,
l’ADN est très étroitement enroulé autour des histones
et l’ARNpolymérase, qui décode l’information génique
et produit l’ARN messager (ARNm), est exclue. La
transcription ne se fait que si la structure de la chroma
tine est ouverte de manière à ce que l’ARNpolymérase
puisse se lier à l’ADN. Pour ce remodeling de la chro
matine, les histones sont modifiées enzymatiquement,
notamment par acétylation (fig. 1). Pour la transcrip
tion des gènes proinflammatoires, il est nécessaire que
des facteurs de transcription proinflammatoires tels
que le NFkB interagissent avec des coactivateurs
capables d’acétyler les segments géniques concernés,
c.àd. qui fonctionnent comme des histoneacétyl
transférases (HAT). Cette transformation est réversible
et catalysée par des histoneacétyldésacétylases
(HDAC). Dans l’asthme par exemple, l’expression des
HAT est augmentée et celle des HDAC diminuée, de
même que leur activité, ce qui favorise la transcription
des gènes proinflammatoires.
Mode d’action moléculaire
des glucocorticoïdes
Le récepteur des glucocorticoïdes (RG) joue un rôle cen
tral dans leur effet. Il n’y a qu’un seul gène pour le RG
humain. La transcription de ce gène, tout comme la
production d’ARNm, sont cependant variables, raison
pour laquelle il y a plusieurs isoformes de RG [1]. Le RG
peut en outre être modifié par accrochage ou largage de
groupes phosphates (phosphorylation, déphosphoryla
tion). Malgré que nous n’ayons qu’un seul gène pour le
RG, il y a de très nombreux produits géniques diffé
rents. Ce qui explique d’une part la grande diversité des
effets biologiques du cortisol et de l’autre la chance que
des effets sélectifs puissent être obtenus par interaction
pharmacologique avec une isoforme particulière du RG.
Le RG se compose de trois différents domaines ou
modules (fig. 2 x), dont l’un se lie au cortisol ou à un
ligand pharmacologique. Un deuxième module peut se
lier à l’ADN. Et le troisième finalement peut interagir
avec d’autres facteurs de transcription et/ou leurs co
facteurs.
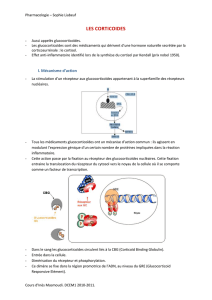
Les mieux connus sont les effets antiinflammatoires
des glucocorticoïdes après leur liaison au RG dans le cy
toplasme de la cellule (fig. 3 x). Cette liaison active le
récepteur et des protéines accessoires, ou chaperons,
s’en détachent. Le RG activé pénètre ensuite dans le
noyau de la cellule. C’est là qu’il déploie son effet bio
logique par régulation de l’expression génique. Cette
dernière peut prendre deux formes. Tout d’abord, deux
RG comme homodimères peuvent se lier directement
à l’ADN, à un dit glucocorticoid-responsive element
(GRE) (fig. 3). L’ expression des gènes peut ainsi être ac
tivée directement ou dans certains cas aussi être in
hibée. L’ important est que de nombreux effets indési
rables des glucocorticoïdes résultent de ce premier
mécanisme. Le nombre de gènes régulés par l’interac
tion directe avec un GRE est relativement petit, et il faut
pour cela des concentrations élevées de glucocorti
coïdes. Fait intéressant, de nombreux gènes activés
dans l’asthme n’ont pas de GRE, malgré qu’ils soient
fortement inhibés par les glucocorticoïdes [2]. Dans
quelle mesure la régulation directe de la transcription
selon le mécanisme présenté est importante pour l’ac
tivité antiinflammatoire des glucocorticoïdes, la ré
ponse à cette question n’est pas assurée.
Un autre argument en faveur du fait que ce mécanisme
a éventuellement une importance secondaire provient
d’un modèle animal. Chez la souris ayant un RG muté,
qui ne peut plus se lier directement à l’ADN, les gluco
corticoïdes ont encore un effet antiinflammatoire très
puissant [7]. Ce qui signifie qu’une bonne partie de leur
effet antiinflammatoire au niveau de la transcription se
AC
AC AC AC
AC
AC
AC
AC AC
AC
AC
AC
HAT
NF-κB ARN
ADN
Histone
Acétylation de
l’histone
Figure 1
Régulation de la transcription par acétylation des histones.
La chromatine est composée d’ADN et d‘histones. Des cofacteurs de transcription interagissent avec des facteurs de transcription tels que
le facteur nucléaire kappa B (NF-κB), qui active l’histone-acétyltransférase (HAT). L’ acétylation des histones qui en résulte ouvre la structure
de la chromatine, ce qui fait que l’ARN-polymérase peut se lier à l’ADN et ensuite initialiser la transcription (modifié d’après [2]).

Forum Med Suisse 2011 ;11(27):473–477 475
curriculum
fait selon un autre mécanisme. Le RG interagit avec
d’autres facteurs de transcription que le NFkB par
exemple, ce qui fait que la transcription de très nom
breux gènes proinflammatoires est inhibée. Dans cet
effet indirect des glucocorticoïdes par l’activité d’autres
facteurs de transcription, c’est notamment l’acétylation
des histones qui est diminuée, ce qui fait que la trans
cription est interrompue. En détail, cela se produit par
la liaison du RG à des cofacteurs de transcription et par
inhibition de leur activité HAT (fig. 3) [2]. Alternative
ment, l’acétylation des histones peut être diminuée et
des histonedésacétylases (HDAC) sont recrutées par le
RG activé (fig. 3). Nul ne sait encore pourquoi les gluco
corticoïdes peuvent interrompre sélectivement la trans
cription de gènes proinflammatoires selon ce méca
nisme. Il est possible que le RG activé se lie en particulier
à des cofacteurs de transcription interagissant avec des
facteurs de transcription proinflammatoires [2].
En plus des effets génomiques des glucocorticoïdes par
la régulation de la transcription, ces derniers peuvent
également agir sur la stabilité de l’ARNm ou exercer leur
effet par interactions directes avec les lipides membra
naires (modification de la fluidité membranaire), des
protéines membranaires (par ex. canaux ioniques) ou
cytoplasmiques [8]. Certains de ces effets sont directe
ment ceux des glucocorticoïdes, d’autres par contre ceux
du RG activé interagissant avec des protéines cytoplas
miques, canaux ioniques ou récepteurs de la membrane
cellulaire. Un effet non génomique peut être très rapide
après action sur les canaux ioniques et la transmission
de signaux intracellulaires (effet sur lesdits second mes-
senger). Les effets génomiques par contre prennent du
temps (5–15 minutes ou même plusieurs heures) en pas
sant par la régulation de la transcription.
U
ballview.org
Récepteur des glucocorticoïdes
(RG)
Liaison du ligand
Liaison d’ADN
Liaison àdes cofacteurs
ADN
ADN
Ligand RG
GRE
A
B
Figure 2
Structure modulaire du récepteur des glucocorticoïdes.
ALe récepteur des glucocorticoïdes (RG) est composé de trois
modules fonctionnels, un pour la liaison du ligand, un pour la
liaison à l’ADN au niveau d‘un glucocorticoid-responsive element
(GRE) et un troisième pour la liaison aux facteurs de transcription
et à leurs cofacteurs.
BModèle détaillé de l’interaction entre les domaines de liaison de
l’ADN et l’ADN (image obtenue avec le logiciel de ballview.org [13]).
U
RG
ADN
UU
GRE
Transcription
ARNm
Membrane
U
RG
HAT
NF-κB HDAC
Désacétylation
-
Noyau
U
RG
STOP
Homodimère
Glucocorticoïdes
Chaperons
12
Figure 3
Mode d’action moléculaire du récepteur des glucocorticoïdes par la régulation de la transcription.
Les glucocorticoïdes traversent la membrane cellulaire et se lient à leur récepteur (RG) dans le cytoplasme, ensuite de quoi des protéines
accessoires (chaperons) se détachent du récepteur. Comme homodimère, le RG se lie à l’ADN nucléaire au niveau du glucocorticoid-
responsive element (GRE), qui régule la transcription de gènes. Alternativement, le RG peut se lier comme monomère à des facteurs de
transcription ou à leurs cofacteurs. Dans ce second mécanisme, le RG inhibe typiquement l’histone-acétylase (HAT) et recrute une histone-
désacétylase (HDAC), ce qui inhibe la transcription.
RG RG

Forum Med Suisse 2011 ;11(27):473–477 476
curriculum
De nombreux effets biologiques des glucocorticoïdes
sont finalement les résultats d’effets génomiques et non
génomiques. Un exemple important est l’effet sur la
mort cellulaire des granulocytes éosinophiles, consi
déré comme central dans leur efficacité dans l’asthme
[9]. Ce qui vaut également pour leur effets immunomo
dulateurs sur les lymphocytes [10].
Résistance aux glucocorticoïdes
Il est notamment bien connu que les patients BPCO réa
gissent mal aux glucocorticoïdes, comparativement aux
asthmatiques. A part cela, il y a des maladies inflamma
toires chroniques telles que l’arthrite rhumatoïde, la
maladie de Crohn ou la colite ulcéreuse, dans lesquelles
les glucocorticoïdes ont un bon effet chez de nombreux
patients, mais pas tous. Plusieurs mécanismes de résis
tance aux glucocorticoïdes sont actuellement connus, et
dans la même maladie plusieurs mécanismes peuvent
intervenir [3]. Nous présentons cidessous à titre
d’exemple un grand mécanisme de la résistance aux
glucocorticoïdes dans la BPCO.
La désacétylation des histones est très nettement dimi
nuée chez les patients BPCO. Ce qui va de pair avec une
activité diminuée de l’HDAC, moins exprimée chez les
patients ayant une grave BPCO et dont l’activité est en
core inhibée sous l’effet du stress oxydatif (fig. 4 x) [2].
Ce qui peut bien expliquer le mauvais effet des gluco
corticoïdes dans la BPCO, car leurs effets antiinflam
matoires sont essentiellement dépendants du recrute
ment des HDAC. De la même manière l’activité des
HDAC peut être massivement réduite chez les fumeurs
asthmatiques (fig. 4), ce qui explique bien pourquoi ils
répondent nettement moins bien aux glucocorticoïdes
topiques ou systémiques.
Perspectives d’optimisation du traitement
par glucocorticoïdes ou modulateurs
non stéroïdiens des glucocorticoïdes
L’ utilisation clinique des glucocorticoïdes est limitée par
leurs effets indésirables dosedépendants. Pour ce qui
est de l’asthme, il est certain que le développement des
stéroïdes topiques a été un jalon, car ils ont surtout un
effet local et les doses de glucocorticoïdes systémiques
ont pu être nettement réduites. Chez l’adulte sous trai
tement systémique prolongé par glucocorticoïdes, les
effets indésirables métaboliques, avec obésité troncu
laire et déclenchement d’un diabète, sans oublier le
risque de manifestation ou d’aggravation d’une ostéo
porose, sont souvent au premier plan. Comme nous
l’avons déjà vu, un autre problème est celui de la résis
tance aux glucocorticoïdes.
Grâce aux progrès dans la connaissance des sites et
mécanismes d’action moléculaires des glucocorticoïdes,
nous pouvons avoir bon espoir de découvrir des ago
nistes sélectifs de leur récepteur ou de leur modulateur,
ayant un bon effet antiinflammatoire et moins d’effets
indésirables. Ces effets résultent souvent de la liaison
directe du RG activé comme homodimère à l’ADN, alors
que par contre l’interaction d’un RG (comme monomère)
avec des facteurs de transcription proinflammatoires
et leurs cofacteurs parvient bien à inhiber l’expression
de nombreuses protéines inflammatoires. Sont particu
lièrement prometteurs les résultats de nouvelles re
cherches avec les modulateurs du RG non stéroïdiens
tels que le Compound A (CpdA) [11]. Le CpdA est une
molécule relativement petite (fig. 5 x) extraite d’une
plante désertique namibienne. L’ effet sélectif ou disso
cié du CpdA peut entre autre s’expliquer par le fait que
le RG par lui activé ne peut pas donner d’homodimère
(fig. 5). Ce RG recrute en outre un autre set de cofac
teurs de transcription. Nous partons actuellement du
principe que de très petites modifications seulement de
la structure du ligand du RG ont une grande influence
sur le profil d’expression génique.
Une autre option futuriste est de vaincre la résistance
aux glucocorticoïdes ou d’en supprimer la cause. Chez
les asthmatiques par exemple, cela est possible par l’ar
rêt de la fumée. Ce qui s’explique par l’absence d’in
hibition des HDAC due à la fumée, ou par la désacé
tylation des histones des gènes proinflammatoires
fonctionnant de nouveau. Fait intéressant la théophyl
line, dont l’effet dans le traitement de l’asthme et de la
BPCO s’explique classiquement par un effet antiinflam
matoire et bronchodilatateur, notamment par inhibi
tion des phosphodiestérases, agit également par une
activation des HDAC. Cet effet de la théophylline a pu
être mis en évidence dans les macrophages alvéolaires
de patients BPCO, dans lesquels à concentrations théra
peutiques (10–5 à 10–6 molaires) la théophylline a multi
plié par 6 l’activité des HDAC. L’ effet de la théophylline
sur les HDAC résulte d’une inhibition de la phospho
Asthme BPCO
Asthme +fumée
NF-κB
NF-κB
U
RG
HDAC↑↑ HDAC↓
Stress oxydatif
Acétylation de
l’histone ↑↑
Acétylation de
l’histone ↓↓
Médiateurs de
l’inflammation ↑↑
Médiateurs de
l’inflammation
Stimulus
Macrophag
ealvéolaire
Figure 4
Modèle de la résistance aux glucocorticoïdes dans la BPCO.
La stimulation des macrophages alvéolaires active des facteurs de transcription
pro-inflammatoires tels que le facteur nucléaire kappa B (NF-κB), ce qui provoque
l’acétylation des histones et ensuite la transcription de gènes pro-inflammatoires.
Les glucocorticoïdes bloquent ce processus en se liant à leur récepteur (RG) et recrutant
l’histone-désacétylase (HDAC), ce qui inhibe l’acétylation induite par le NF-κB et
l’activation des gènes pro-inflammatoires. Chez les patients BPCO ou asthmatiques
fumeurs, l’activité de l’HDAC est nettement diminuée sous l’effet du stress oxydatif,
ce qui augmente l’activité du NF-κB et diminue la réponse aux glucocorticoïdes, c.-à-d.
qu’il en résulte une résistance aux glucocorticoïdes (modifié d’après [2]).

Forum Med Suisse 2011 ;11(27):473–477 477
curriculum
inositolkinase 3 delta (PI3Kd) [3, 12]. Pour cette raison,
les inhibiteurs sélectifs de la PI3Kdsont potentiellement
de nouveaux médicaments permettant de vaincre la
résistance aux glucocorticoïdes.
En résumé, les toutes dernières connaissances sur les
mécanismes d’action moléculaires des glucocorticoïdes
non seulement sont très intéressantes mais ouvrent dif
férentes options d’améliorer encore le traitement de
pathologies inflammatoires et éventuellement autres
maladies par modulation de leur effet et interaction
(plus) sélective avec le RG. Nous n’en sommes proba
blement qu’au début d’une histoire à succès dont la fin
influencera sans doute le traitement de beaucoup de
nos patients.
Remerciements
L’ auteur remercie les collègues cidessous de leur lecture attentive du
manuscrit, de leurs suggestions et discussions: Madame la Prof. B. C.
Biedermann (cabinet médical à 8345 Adetswil), Madame Dr C. Rosamilia
(médecinassistante, Medizinische Universitätsklinik, Kantonsspital
Bruderholz) et Monsieur le Prof. S. Krähenbühl (Médecinchef, Abteilung
Klinische Pharmakologie & Toxikologie, Universitätsspital Basel).
Correspondance:
Dr Andreas W. Jehle
Transplantationsimmunologie und Nephrologie
Universitätsspital Basel
CH-4031 Basel
Références recommandées
–Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids –
new mechanisms for old drugs. N Engl J Med. 2005;353(16):1711–23.
–Barnes PJ. How corticosteroids control inflammation: Quintiles Prize
Lecture 2005. Br J Pharmacol. 2006;148(3):245–54.
–Reichardt HM, et al. Repression of inflammatory responses in the
absence of DNA binding by the glucocorticoid receptor. Embo J.
2001;20(24):7168–73.
–Druilhe A, Letuve S, Pretolani M. Glucocorticoidinduced apoptosis
in human eosinophils: mechanisms of action. Apoptosis. 2003;8(5):
481–95.
–De Bosscher K, Haegeman G, Elewaut D. Targeting inflammation
using selective glucocorticoid receptor modulators. Curr Opin Pharma
col. 2010;10(4):497–504.
Vous trouverez la liste complète et numérotée des références
dans la version en ligne de cet article sous www.medicalforum.ch.
CH CH2NH2Cl
CH3
Cl
CH3C
O
O
Modulateur non stéroïdien
du RG CpdA
Agoniste stéroïdien du RG
Prednisone
Effet (plus) sélectif
AB
ADN ADN
Figure 5
Régulation différenciée de la transcription par modulateurs sélectifs du récepteur des glucocorticoïdes.
ALes ligands classiques du récepteur des glucocorticoïdes (RG) tels que la prednisone régulent la transcription selon deux mécanismes
différents. D’une part, la prednisone provoque une homodimérisation du RG avec liaison consécutive de l’homodimère à un glucocorticoid-
responsive element (GRE). Le RG lié à la prednisone peut d’autre part interagir avec d’autres facteurs de transcription ou leurs cofacteurs.
Alors que le premier mécanisme est responsable de nombreux effets indésirables métaboliques des glucocorticoïdes, le second inhibe
des facteurs de transcription pro-inflammatoires tels que le facteur nucléaire kappa B (NF-κB) par inhibition de l’histone-acétylase (HAT) et
recrutement de l’histone-désacétylase (HDAC).
BDe tout nouveaux modulateurs du récepteur des glucocorticoïdes, dont certains non stéroïdiens tels que le Compound A (CpdA),
empêchent toute homodimérisation du RG et agissent notamment par inhibition de l’HAT et recrutement de l’HDAC, ce qui explique
le meilleur profil d’effets/effets indésirables du CpdA.
Noyeau Noyeau
Pas de dimérisation
Activation
Activation
3Pas de liaison
au GRE
Membrane cellulaire
Membrane cellulaire
Transcription RG RG
RG
RG
RG
RG
RG
RG
RG RG
 6
6
1
/
6
100%