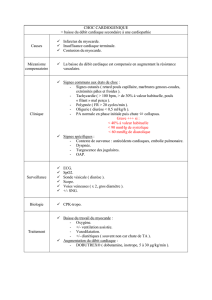
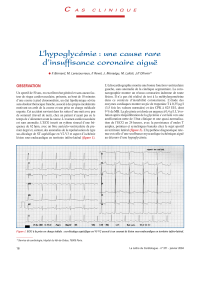
la fonction contractile regionale du myocarde post

Université Paris XII Val de Marne
Ecole Doctorale Sciences de la Vie et de la Santé
Doctorat de Pharmacologie Expérimentale et Clinique
Laurence LUCATS
LA FONCTION CONTRACTILE REGIONALE DU
MYOCARDE POST-ISCHEMIQUE :
ADAPTATIONS PHYSIOPATHOLOGIQUES ET
PHARMACOLOGIQUES
Soutenue le 15 décembre 2006
Membres du Jury :
Monsieur le Professeur Alain Berdeaux Président
Madame le Professeur Valérie Chetboul Rapporteur
Monsieur le Professeur Jean-Jacques Mercadier Rapporteur
Madame le Professeur Geneviève Derumeaux Examinateur
Monsieur le Professeur Guy Heyndrickx Examinateur
Monsieur le Docteur Bijan Ghaleh-Marzban Directeur

A ma Famille,
A mes Amis,
Alice & Olivier, Claire, Soline & Olivier, Etienne, Caroline & Bruno, Cécile &
Bernard, Aude, Marie-Agnès & Olivier, Elizabeth, Guillaume
Pour nos bons moments et vos encouragements,
Grâce aux quels, je ne suis pas El Desdichado.

A Monsieur le Professeur Alain Berdeaux,
Qui m’a ouvert les portes de son laboratoire,
Qui m’a fait profiter chaque jour de sa grande culture scientifique
Et qui me fait l’honneur de présider le jury de cette thèse.
A Madame le Professeur Valérie Chetboul,
Dont je suis fière d’avoir été l’élève pendant mes études vétérinaires,
Qui m’a fait profiter de ses talents d’échocardiographiste pour mon travail de
thèse,
Et qui m’honore aujourd’hui en étant le rapporteur de ce travail.
A Monsieur le Professeur Jean-Jacques Mercadier,
Qui nous a fait profiter de sa maîtrise et de son savoir pour l’analyse et l’interprétation
coordonnées des résultats de physiologie intégrée et de biologie moléculaire,
Et qui me fait le grand honneur d’être rapporteur de ce travail.
A Madame le Professeur Geneviève Derumeaux,
Qui me fait le grand honneur d’être examinateur de ce travail.
A Monsieur le Professeur Guy Heyndrickx,
Qui me fait le grand honneur d’être examinateur de ce travail.
A Monsieur le Docteur Bijan Ghaleh,
Qui m’a appris chacune des étapes de la recherche avec précaution et rigueur,
De l’élaboration de l’étude à la réponse au reviewing, merci.
A Monsieur Alain Bizé,
Qui m’a offert une aide constante et infaillible, toutes ses petites manies,
minuties et précautions ont fait de lui un soutien indispensable.

2
A Monsieur le Docteur Renaud Tissier,
Qui m’a accompagnée pendant ces quatre années au laboratoire,
Qui, en plus de son contribution scientifique, a apporté à notre équipe ses
qualités humaines exceptionnelles. Son bagout et sa bonne humeur nous ont soutenus
de l’analyse scientifique aux petites réjouissances du quotidien,
Qu’il soit assuré de ma plus grande admiration et de ma sympathie dévouée.
A Madame le Docteur Virginie Chalvignac et Messieurs les Docteurs Nicolas Couvreur,
Dominique Tessier et Xavier Waintraub,
Avec qui j’ai eu le plaisir de travailler.
A Messieurs les Docteurs Patrice Colin et Xavier Monnet,
Qui ont apporté à ces travaux leur expérience et leur esprit critique.
A Madame le Docteur Patricia Rouet-Benizeb et Monsieur Laurent Vinet,
Pour l’experte collaboration qu’ils m’ont offerte.
A Monsieur le Docteur Thibaud Damy,
Qui m’a fait part de son grand savoir en matière de NOS,
Sa sympathie, son écoute et ses conseils m’ont portée dans les moments
difficiles de ce travail.
A Messieurs les Docteurs Didier Morin, Jinbo Su et Roland Zini,
Pour le précieux savoir-faire qu’ils ont apporté à ce travail.
A Monsieur le Professeur Luc Hittinger,
Dont l’acuité scientifique nous a aidés dans l’analyse ces travaux,
Et dont les corrections ont amélioré nos manuscrits.

3
A Madame le Professeur Brigitte Enriquez,
Qui m’a initiée à la pharmacologie,
Pour sa gentillesse, son ouverture et son attention aux autres.
A Monsieur le Professeur Jean-Louis Pouchelon,
Pour l’intérêt qu’il a porté à mon travail.
A l’ensemble de l’Unité de Cardiologie d’Alfort ; Vassiliki Gouni, Audrey Niccole,
Carolina Carlos et François Serres,
Pour leur aide attentionnée lors des examens échocardiographiques de ce
travail.
A Jean-Paul Vilaine et à l’ensemble de l’équipe des « Biological studies » du
Laboratoire Servier : Florence Mahlberg-Gaudin, Pascale Gluais, Muriel Bouly et Romain Bos,
Qui nous ont fait part de leur expérience de l’ivabradine.
A Monsieur Georges Zadigue,
Pour le soin qu’il a apporté aux animaux,
Pour son aide généreuse, son altruisme et sa sérénité.
A Monsieur le Professeur Jean-François Giudicelli,
Qui m’a accueilli au sein du Laboratoire de Pharmacologie de Bicêtre,
Qui a accompagné le commencement de ce travail,
Et qui m’a laissé entrevoir l’étendue de son impressionnante culture.
A Madame le Docteur Christine Richer,
Pour les aimables discussions et l’attention qu’elle a portée à mon travail.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
1
/
191
100%