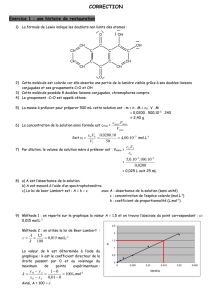
CORRECTION TD Chapitre 3

CORRECTION TD Chapitre 3 :
Evolution temporelle d'un système : vitesses de réaction et lois de vitesse
Exercice 1 : Décomposition du bromure de nitrosyle : (tt en 1 p228)
La réaction de décomposition du bromure de nitrosyle en phase gazeuse :
NOBr(g) → NO (g) + ½ Br2 (g)
suit la loi de vitesse : v=k[NOBr]2.
Temps (s) 0 6,2 10,8 14,7 20 24,6
[NOBr] (mol.L-1) 0,0250 0,0191 0,0162 0,0144 0,0125 0,0112
1/[NOBr] 40,0000 52,3560 61,73 69,4444 80,0000 89,2857
→ Vérifier que la réaction est bien du deuxième ordre par rapport au bromure de nitrosyle et
déterminer la constante de vitesse de la réaction.
Si la réaction est bien d'ordre 2, la concentration au cours du temps suit cette équation :
1
[NOBr](t)=1
[NOBr]0
+k t
Il faut donc tracer
1
[NOBr](t)=f(t)
On a une belle droite, donc la loi est vérifiée. le coefficient directeur permet d'accéder directement
à la valeur de k :
k=2,003 mol-1.L.s-1.
Pour réinvestir : Détermination d'une loi de vitesse : décomposition de l'azométhane en phase
gazeuse : (tout en 1 p247)
Dans un récipient de volume fixé, on introduit à 600K de l'azométhane. Celui-ci se décompose en
éthane et en diazote suivant l'équation bilan :
CH3N2CH3 (g) = CH3CH3 (g) + N2(g)
L'évolution de la réaction est suivie par manométrie, et une série de mesures a donné la pression
partielle pA en azométhane :
t (103s) 0 1,00 2,00 3,00 4,00
pA (10-2 mmHg) p0=8,21 5,74 4,00 2,80 1,96
→ Vérifier que la réaction est d'ordre 1 par rapport au réactif et calculer sa constante de vitesse. On
supposera que tous les gaz ou mélanges de gaz sont parfaits.
Même principe avec la loi de vitesse :
ln [A](t)=ln [A]0−ν k t
Ici d'après la loi des gaz parfaits :
p0=[CH 3N2CH 3]0RT
et
pA=[CH 3N2CH 3]tRT

la relation devient :
ln([A](t)
[A]0
)=−kt
soit
ln(pA
p0
)=−kt
On trace
ln(pA
p0
)= f(t)
, on devrait avoir une droite décroissante de pente k :
d'où
k=0,358 .10−3s−1
(attention le temps dans le tableau est en 103s …)
Exercice 2 : Datation au carbone 14 : (tt en 1p231)
La période de décomposition radioactive du carbone 14 vaut τ=5730ans. Un échantillon
archéologique contient un morceau de bois mort contenant 74% du carbone 14 contenu usuellement
dans la matière vivante.
14
7C → 14
7N+0
−1e
1- Sachant que la réaction est d'ordre 1, établir la loi de vitesse et l'expression du temps de demi-
réaction.
ln [C](t)=ln[C]0−k t
et
t1/2=ln 2
k
2- En déduire la valeur de k
k=ln 2
t1/2
=ln 2
5730 =1,21.10−4an−1
3- Depuis combien de temps l'arbre est-il mort ?
soit tM la durée depuis la mort, on sait que
[C]=0,74[C]0
→
ln([C](t)
[C]0
)=ln 0,74=−k tM
d'où
tM=−ln 0,74
1,21.10−4=2488 ans
Pour réinvestir : Utilisation du soufre radioactif comme traceur biologique : (tout en 1 p248)
L'isotope 38S est radioactif avec une période (ou durée de demi-vie) de τ=2,84h ; il est utilisé pour
étudier le métabolisme des protéines. On considère un échantillon de protéine marqué par l'isotope
38S ; cet échantillon présente une activité initiale de 48000 désintégrations par minutes.
→ Calculer l'activité de cet échantillon au bout de 8h et au bout de 24h sachant que la décroissance
radioactive suit une loi cinétique d'ordre 1.
L'activité est directement proportionnel au nombre de noyaux radioactif, comme c'est une loi
d'ordre 1 on peut écrire :
A(t)=A0exp(−k t)
k la constante de vitesse est liée au temps de demi-réaction,soit la durée de demi-vie par la formule :

k=ln 2
τ
L'expression de la décroissance radioactive devient :
A(t)=A0exp (−t ln2
τ)
Application numérique :
au bout de 8h :
A(8)=48000 exp(−8×ln2
2,84 )=6812 desintegration /min
au bout de 24h :
A(24)=48000 exp(−24×ln2
2,84 )=137 desintegration /min
Exercice 3 : Décomposition du monoxyde d'azote (tout en 1 p249)
Dans cet exercice, on suppose que les mélanges gazeux se comportant comme des mélanges parfaits
de gaz parfaits. La décomposition à 1151°C de l'oxyde nitreux a lieu suivant la réaction :
2NO(g) → N2(g) + O2(g)
A volume constant et pour une pression initiale d'oxyde nitrique p0=200mHg, la pression partielle p
de NO varie en fonction du temps de la manière suivante :
p (mmHg) 200 156 128 108 94 83
t (min) 0 5 10 15 20 25
Dans les mêmes conditions, mais pour des pressions initiales de NO différentes, on a déterminé les
vitesses initiales de disparition du monoxyde d'azote correspondantes, notées v0 :
p0 (mmHg) 100 150 200 300 400
vNO,0 (mmHg.min-1) 2,8 6 11 25 45
1- Déterminer l'ordre de la réaction en vous basant sur les valeurs v0.
la loi de vitesse s'écrit :
Attention on a les mesures de la vitesse de disparition du monoxyde de diazote, que l'on notera vNO,
à ne pas confondre avec la vitesse globale de réaction que l'on notera v.
v=−1
2
d[NO]
dt =k[NO ]n
vNO =−d[NO]
dt =2k [NO ]n=k ' pNO
n
avec
k '=2k
(RT )n
On linéarise la loi de vitesse :
ln(vNO )=ln (k ')+nln (pNO )
On trace ln(vNO) en fonction de ln(pNO), on devrait avoir une droite
de pente n.
→ On trouve bien une pente de coefficient directeur 2,01 → n = 2
2- Ecrire et intégrer l'équation cinétique.
Comme c'est une loi d'ordre 2 :
d[NO]
dt =−2k [NO ]2
pNO =RT [NO ]→1
RT
d pNO
dt =−2k
(RT )2pNO
2
→
d pNO
dt =−2k
RT pNO
2
en intégrant on obtient :
1
pNO
=2kt
RT +1
PNO ,0

3- Vérifier l'ordre obtenu en utilisant une méthode graphique avec les valeurs du tableau 1.
On trace 1/pNO = f(t) → c'et une droite, l'ordre est validé.
4- Une étude en fonction de la température a donné les résultats suivants :
θ (°C) 974 1057 1260
2k (mol-1.mL.s-1) 20,5 87 2100
Evaluer graphiquement l'énergie d'activation et déterminer le facteur préexponentiel.
loi d'Arrhénius :
k=Aexp(−Ea
RT )
forme linéaire :
ln k=ln A – Ea
RT
: on trace ln(k) = f(1/T), le coefficient directeur donnera accès à
Ea et l'ordonnée à l'origine à A :
Ea
R=3,11.104→ Ea=3,11 .104×8,31=258 kJ.mol−1
ln A = 27,2 →
A=exp(27,2)=6,50.1011 mol−1.mL.s−1
Donnée : R=8,31 J.K-1.mol-1=62,3 mmHg.L.mol-1.K-1
Exercice 4 : Temps de demi-réaction : (violet p49)
En solution dans l'éthanol, la potasse KOH est totalement dissociée. On étudie à 20°C sa réaction
avec le 1-bromo-2-méthylpropane (noté RBr) qui conduit au 2-méthylpropan-1-ol (noté ROH) par
substitution.
1- Ecrire l'équation bilan de la réaction.
Il s'agit d'une substitution de Br- par HO- : R-Br + HO- → R-OH + Br-
2- Définir le temps de demi-réaction. Dans le cas d'une réaction A → B d'ordre n, exprimer t1/2 en
fonction de la constante de vitesse k et de [A0] pour n=0,1ou2.
Le temps de demi-réaction est la durée nécessaire pour atteindre la moitié de l'avancement final.
Voir cours pour la démonstration :
n = 0 :
t1/2=[A]0
2k
n = 1 :
t1/2=ln 2
k
n = 2 :
t1/2=1
k[A0]
3- Une première expérience a pour conditions initiales :
[Rbr ]0=1,00 .10−2mol.L−1
et
[HO−]0=1,00 mol.L−1
.
On détermine la concentration de RBr à l'instant t :
t (min) 0 10 20 30 40
102 [RBr] 1,00 0,50 0,25 0,12 0,06

a- Comment peut-on déterminer [RBr]t ?
Comme la réaction fait intervenir des ions, on peut utiliser la conductimétrie.
b- Pourquoi utiliser des concentrations si différentes en réactifs ?
→ Dégénéresence de l'ordre : comme [HO-]>> [Rbr], la concentration en [HO-] variera très peu
durant la réaction, on peut considérer que [HO-] ≈ [HO-]0
la loi de vitesse s'écrit donc :
v=k[RBr]n1[HO−]n2=kapp [RBr]n1
avec
kapp=k[HO−]0
n2
(constante de vitesse apparente).
c- Déterminer à l'aide du tableau numérique trois valeurs de t1/2 à différentes origines. Cette réaction
admet-elle un ordre ? Si oui, quel est-il et combien vaut la constante de vitesse apparent ?
Expérience 1 : on part de t=0min : on voit que t1/2=10min (la moitié de la quantité initiale à réagit).
Expérience 2 : on part de t=10min → t1/2 = 10min
Expérience 3 : on part de t=20min → t1/2 = 10 min
On en déduit que le temps de demi-réaction est indépendant de la concentration initiale en Rbr : la
réaction est donc d'ordre 1 par rapport à RBr. → n1 = 1
kapp=ln 2
t1/2
=6,9 .10−2min−1
4- On recommence la même expérience avec les conditions initiales
[RBr]0=1,00.10−2mol.L−1
et
[HO−]0=0,50 mol.L−1
.
t (min) 0 10 20 30 40
102 [RBr] 1,00 0,71 0,50 0,35 0,25
a- Déterminer des valeurs de t1/2 et en déduire éventuellement une constante apparente de vitesse.
b- En déduire l'ordre partiel par rapport à HO-. Donner la loi de vitesse générale.
Dans cette situation, t1/2 = 20min
kapp=ln 2
t1/2
=3,5.10−2min−1
Etant donné que lorsqu'on divise la concentration initiale en [HO-] la constante de vitesse apparente
est divisée par 2 également cela impose n2=1 (car cela traduit le fait que kapp est proportionnelle à
la concentration).
La loi de vitesse générale s'écrit donc :
v=k[RBr][ HO−]
avec
k=kapp1
[HO−]0,1
=6,9.10−2
1,00 =6,9 .10−2L.mol−1.min−1
Exercice 5 : Efficacité de la trempe thermique (tout en1 p243)
La cinétique d'une réaction chimique est suivie au moyen d'une méthode d'analyse chimique
précédée d'une trempe par refroidissement de 100°C (température de la réaction) à 0°C (température
du dosage). En considérant que cette réaction possède une énergie d'activation Ea=40 kJ.mol-1,
quelle est l'efficacité de la trempe par refroidissement ?
Si l'énergie d'activation est de 80kJ.mol-1, l'efficacité de la trempe est-elle meilleure ou moindre ?
 6
7
6
7
1
/
7
100%