correction. - Enseignements informatiques et mathématiques en

Module LV348 Version enseignant TD 1 – page 1/7
TD 1 Biais de codons
Revu dernièrement par Mathilde Carpentier, Cyril Gallut et Joël Pothier
Version du 15 janvier 2014
L’objectif de ce TP est de prendre en main le langage python et de revoir quelques notions fonda-
mentales sur l’ADN (complémentarité, phase de lecture, traduction et usage du code génétique.
La question biologique posée porte sur l’usage du code génétique.
1 Les données
Nous allons travailler sur le génome complet de la bactérie Escherichia coli. Nous allons téléchar-
ger son génome complet sur le site ftp du NCBI ftp://ftp.ncbi.nlm.nih.gov/.
Ce serveur propose les génomes de très nombreux organismes. Il y a en effet maintenant environ
1000 génomes complets séquencés, et des millions de séquences dispersées. Différents fichiers sont
proposés pour chaque génome, ils correspondent à différents formats des données. Nous allons utiliser
le fichier .fna, c’est-à-dire la séquence nucléique du génome complet au format fasta. L’adresse du fi-
chier est : ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/Escherichia_coli_
K_12_substr__MG1655/NC_000913.fna 1
Le format fasta est un format texte pour les séquences nucléiques (ADN) ou protéiques. Une
séquence de ce format comporte une ligne de description commençant par un ">" puis les lignes de
la séquence. La fin d’une séquence est atteinte lorsque la fin du fichier est atteinte ou lorsqu’une autre
séquence commence, c’est-à-dire que l’on rencontre une ligne commençant par un ">". La ligne de
description comporte un identifiant de la séquence dans la banque d’où elle provient.
Exemple de séquence au format fasta :
>NC_010473|:163816-163951|small RNA| [gene=tff] [locus_tag=ECDH10B_0148]
CGGACTTCCGATCCATTTCGTATACACAGACTGGACGGAAGCGACAATCTCACTTTGTGTAACAACACAC
ACGTATCGGCACATATTCCGGGGTGCCCTTTGGGGTCGGTAATATGGGATACGTGGAGGCATAACC
2 Exercices
Tous les exercices sont à faire en python.
2.1 Lecture des fichiers
Ecrivez une fonction python permettant de lire un fichier fasta ne contenant qu’une séquence et
qui retourne la séquence lue sous la forme d’une chaî,e de caractères..
Solution :
Présentation de python : fonction, for, ouverture, fermeture et lecture de fichier.
1. Il vaut mieux ne pas prendre ...Escherichia_coli_K_12_substr__DH10B/NC_010473.fna car elle
contient un Y
c
2013-2014 (by UPMC/Licence de biologie/LV348) 15 janvier 2014

Module LV348 Version enseignant TD 1 – page 2/7
1# lecture d’un fichier au format fasta
2# version 1: la sequence est
3#recuperee sous la forme d’une chaine de caracteres
4#------------------------------------------------------------------------
5def readFasta(file):
6infile=open(file, ’r’)
7seq=""
8name = ""
9tmp=infile.readlines()
10 infile.close()
11
12 #name =tmp[0][1:-1] # pour eliminer le > du debut et le \n de fin
13 for xin tmp[1:] :
14 seq=seq+x[:-1]
15
16 #return (name,seq) # la fonction retourne le nom et la sequence
17 return seq # la fonction retourne la sequence
./code/lecture.py
2.2 Le brin complémentaire
Comme vous l’avez peut être remarqué, il n’y a qu’un seul des deux brins d’ADN dans le fi-
chier fasta. Ecrivez une fonction python retournant le brin complémentaire d’une séquence nucléique
donnée en argument.
Solution :
Python : while et dictionnaire
Bio : complémentarité des brins
1# donne le complementaire inverse de la sequence seq
2def complement(seq):
3compl = {’A’:’T’,’C’:’G’,’G’:’C’,’T’:’A’}
4cseq=’’
5i = len(seq) -1
6while i>=0:
7cseq = cseq + compl.get(seq[i], ’N’)
8i -= 1
9return cseq
./code/complementaire.py
2.3 Détection des CDS
Ecrivez une fonction python qui retourne la liste de toutes les sous séquences commençant par le
codon "start" et se terminant par un codon "stop" en phase (n’oubliez pas le brin complémentaire).
Ces séquences sont nommées CDS (Coding sequences).
Note : Si plusieurs start sont trouvés dans la même phase de lecture avant un stop, le CDS débute au
premier start ; les CDS peuvent se chevaucher (sur différentes phases, et éventuellement sur le brin
d’ADN complémentaire),
c
2013-2014 (by UPMC/Licence de biologie/LV348) 15 janvier 2014

Module LV348 Version enseignant TD 1 – page 3/7
Solution :
Encore des dico, introduction des autres types énumérés : liste et tuple .
Question plus bio : les phases de lectures, détection des CDS.
Pour info, les ORFs sont les séquences entre 2 STOP en phase et non un START et un STOP
C’est le premier algo un peu compliqué, bien y réfléchir.
1# Retourne la liste des orf de la seq
2#
3def findorf(seq):
4start = ("ATG")
5stop = ("TAA","TAG","TGA")
6startlist = [-1,-1,-1]
7orflist = []
8for iin range(len(seq)-2):
9if seq[i:i+3] in start:
10 if startlist[i%3] == -1:
11 startlist[i%3] = i
12 elif seq[i:i+3] in stop:
13 if startlist[i%3] != -1:
14 # Attention n’inclus pas le stop
15 orflist.append(seq[startlist[i%3]:i])
16 startlist[i%3] = -1
17 return orflist
./code/orf.py
2.4 Traduction
Ecrivez une fonction python qui à partir d’une séquence nucléique retourne la séquence protéique
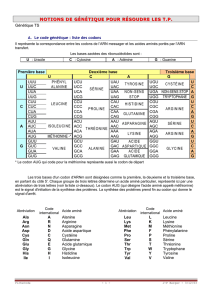
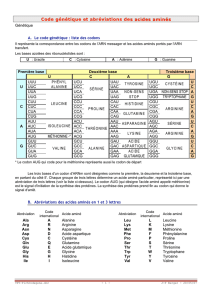
correspondante (en utilisant le code génétique à une lettre qui vous a été donné et qui se trouve au lien
suivant : http://www.snv.jussieu.fr/bmedia/codegenet/CodeGenet.htm).
Solution :
Cette fonction ne nous servira pas vraiment mais elle permet d’introduire le code génétique et de
parler de redondance.
1# effectue la traduction de la sequence seq
2def traduction(seq):
3code = {"TTT":"F","TTC":"F","TTA":"L","TTG":"L","TCT":"S","TCC":"S",
4"TCA":"S","TCG":"S","TAT":"Y","TAC":"Y","TAA":"*","TAG":"*",
5"TGT":"C","TGC":"C","TGA":"*","TGG":"W","CTT":"L","CTC":"L",
6"CTA":"L","CTG":"L","CCT":"P","CCC":"P","CCA":"P","CCG":"P",
7"CAT":"H","CAC":"H","CAA":"Q","CAG":"Q","CGT":"R","CGC":"R",
8"CGA":"R","CGG":"R","ATT":"I","ATC":"I","ATA":"I","ATG":"M",
9"ACT":"T","ACC":"T","ACA":"T","ACG":"T","AAT":"N","AAC":"N",
10 "AAA":"K","AAG":"K","AGT":"S","AGC":"S","AGA":"R","AGG":"R",
11 "GTT":"V","GTC":"V","GTA":"V","GTG":"V","GCT":"A","GCC":"A",
12 "GCA":"A","GCG":"A","GAT":"D","GAC":"D","GAA":"E","GAG":"E",
13 "GGT":"G","GGC":"G","GGA":"G","GGG":"G"}
14 tradseq = ’’
15 for iin range(0,len(seq)-2,3):
16 tradseq += code.get(seq[i:i+3], ’X’)
c
2013-2014 (by UPMC/Licence de biologie/LV348) 15 janvier 2014

Module LV348 Version enseignant TD 1 – page 4/7
17 return tradseq
./code/traduction.py
2.5 Usage des codons
Ecrivez un programme python qui calcule la fréquence d’utilisation de chaque codon pour chaque
acide aminé d’une liste de séquences nucléiques donnée en argument. Appliquez là aux CDS détec-
tées.
Par exemple, l’usage des codons pour les cds détectées sur le génome d’E. coli que vous avez
téléchargé est
A GCA: 22.47% GCC: 26.81% GCG: 33.03% GCT: 17.70%
Solution :
1# calcul la frequence de chaque codon par acide amine
2def calcul_freq_codon_aa(lesSeq):
3freq_par_aa = {
4"A":{"GCT":0,"GCC":0,"GCA":0,"GCG":0},
5"C":{"TGT":0,"TGC":0},
6"D":{"GAT":0,"GAC":0},
7"E":{"GAA":0,"GAG":0},
8"F":{"TTT":0,"TTC":0},
9"G":{"GGT":0,"GGC":0,"GGA":0,"GGG":0},
10 "H":{"CAT":0,"CAC":0},
11 "I":{"ATT":0,"ATC":0,"ATA":0},
12 "K":{"AAA":0,"AAG":0},
13 "L":{"CTT":0,"CTC":0,"CTA":0,"CTG":0,"TTA":0,"TTG":0},
14 "M":{"ATG":0},
15 "N":{"AAT":0,"AAC":0},
16 "P":{"CCT":0,"CCC":0,"CCA":0,"CCG":0},
17 "Q":{"CAA":0,"CAG":0},
18 "R":{"AGA":0,"AGG":0},
19 "R":{"CGT":0,"CGC":0,"CGA":0,"CGG":0},
20 "S":{"TCT":0,"TCC":0,"TCA":0,"TCG":0,"AGT":0,"AGC":0},
21 "T":{"ACT":0,"ACC":0,"ACA":0,"ACG":0},
22 "V":{"GTT":0,"GTC":0,"GTA":0,"GTG":0},
23 "W":{"TGG":0},
24 "Y":{"TAT":0,"TAC":0}}
25 #1) compter les codons
26 codon={}
27 for sin lesSeq:
28 for iin range(0,len(s)-2,3):
29 if s[i:i+3] in codon :
30 codon[s[i:i+3]]+=1
31 else :
32 codon[s[i:i+3]]=1
33 #2) Calculer les frequences par aa
34 for aa in freq_par_aa :
c
2013-2014 (by UPMC/Licence de biologie/LV348) 15 janvier 2014

Module LV348 Version enseignant TD 1 – page 5/7
35 freqtotal = 0.0
36 # calcul la frequence cumulee des codon par aa pour normaliser
37 for cod in freq_par_aa[aa] :
38 freqtotal += codon[cod]
39 # calcul la frequence de chaque codon par acide amine
40 for cod in freq_par_aa[aa] :
41 freq_par_aa[aa][cod] = (codon[cod]/freqtotal)*100
42 return freq_par_aa
./code/usage_codon.py
3 Bonus
Ecrivez un programme python qui calcul le Nc(effective number of codons) pour les CDS détec-
tés.
Ecrivez un programme python qui calcul le CAI (codon adaptation index) pour les CDS détectés.
Comparez les valeurs.
3.1 Conclusion
Observez la distribution globale des codons, la composition globale des séquences en acides ami-
nés (faire des graphiques à l’aide de gnuplot ou d’un tableur). Sachant que le code génétique est
redondant, nous voulons analyser si les codons codant pour un même acide aminé sont utilisés de ma-
nière aléatoire ou non. Calculez aussi l’usage global des codons pour chaque acide aminé, c’est-à-dire
le pourcentage de chaque codon possible pour un acide aminé. L’usage des codons vous semble-t-il
uniforme pour chaque acides aminé ? Question subsidiaire : comment vérifieriez-vous statistiquement
cette hypothèse de non-uniformité ? Faites le si possible.
Pour conclure, il est conseillé de lire la page : http://fr.wikipedia.org/wiki/Biais_
d’usage_du_code
Solution :
Toutes les fonctions python sont dans TD1_Corrections.py dans le répertoire code
1#!/usr/bin/python
2# -*- coding: utf-8 -*-
3
4import getopt
5import sys # permet la lecture des arguments de la ligne de commande.
6import math # permet d’utiliser les fonctions math (pow)
7
8from ORF import *
9from complementaire import *
10 from compoAA import *
11 from compte_codon import *
12 from lecture import *
13 from traduction import *
14 from usage_codon import *
15
c
2013-2014 (by UPMC/Licence de biologie/LV348) 15 janvier 2014
 6
7
6
7
1
/
7
100%