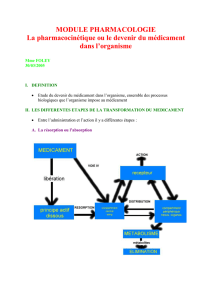
Resorption - Absorption - Effets de 1er passage

I. Résorption / Absorption
Site d’application → circulation générale (par un temps de latence)
1) Passage des membranes
1) Caractéristiques
Définitions
- Processus par lequel le principe actif ou une prodrogue passe de son site d’application dans le flux
sanguin
- Processus complexe :
o Pénétration dans les structures membranaires
o Pénétration au travers de structures dans le milieu externe et les tissus interstitiels
o Résorption dans le milieu sanguin
Absorption = phase de résorption + effets de 1er passage
Résorption différente selon la voie d’administration
- Tractus Gastro-Intestinale → administration orale
- Tissus sous-cutané → administration sous-cutanée (résorption variable selon dilatation des vaisseaux
sanguins)
- Muscle → administration Intra-Musculaire (résorption rapide car richesse des vaisseaux sanguins)
- Muqueuses :
o Rectale
o Pulmonaire (muqueuse alvéolaire)
o Nasale
o Bucco-linguale
La diffusion dépendra de la vascularisation, de la structure des membranes
- Rappel physiologique du tube digestif :
o pH de l’estomac = 2
o pH de l’intestin grêle = 6 – 7
o pH du gros intestin = 8
o pH du sang se situe entre 7.32 et 7.42
Caractéristiques de la membrane Gastro-Intestinale
- Double-couche lipidique recouverte d’une monocouche de protéines
- Protéines extrinsèques en surface
- Protéines intrinsèques = rôle dans les échanges de substances hydrosolubles
2) Mécanismes d’absorption
- Diffusion passive : transport paracellulaire, transporteur cellulaire

- Diffusion facilité : passage des molécules dans le sens d’un gradient de concentration, avec une vitesse
supérieure à celle d’un phénomène de diffusion passive.
- Transport actif : ATP dépendant
Transport actif et diffusion facilitée
- Transporteur d’influx : solute carriers SLC
o OATP (organic anion transporter peptides) pH dépendant
o OCT : organic cation transporter
o OAT : organic anion transporter
o PepT1 : co-transport H+
Transporteurs ABC (ATP Binding Cassette) : MRP 1, MRP3, MRP5
- Transporteur d’efflux ATP dépendants
o Transporteurs ABC :
MDR (multi-drug resistanceà ou Pgp (P-glycoprotéine)
MRP (multi-resistance protein) : MRP2, MRP4, MRP6
BCRP (Breast Cancer resistance protein) ou MXR
3) Conditions de franchissement
1) Caractéristiques liées au médicament = propriétés physico-chimiques
a) Hydrosolubilité
Médicament est absorbé seulement sous forme dissoute dans le tube digestif
b) Nature chimique, pKa et état d’ionisation
Dans le tractus GI, le médicament existe sous forme ionisée et non ionisée
Etat d’ionisation est un facteur déterminant de l’absorption car seule la forme non
ionisée du médicament est absorbée
La répartition des formes non ionisées et ionisées dépend du pH du milieu et du pKa du composé
Permet de connaître le niveau d’absorption d’un médicament au niveau stomacal ou
duodénal
Médicaments = acides ou bases faibles qui se dissocient
Le pKa (50% de dissociation) caractérise chaque substance
L’équation de Henderson-Hasselbach permet de déterminer la fraction non ionisée du composé en fonction du
pH du milieu et du pKa du médicament.
- Acide : pH = pKa + Log ( Ci / Cni )
- Base : pH = pKa + Log ( Cni / Ci )
A un pKa bas correspond un acide fort ou une base faible
Quand 2 formes sont en équilibre : pH = pKa → 50% forme ionisée (Log 1 = 0)
A pH + 1 = pKa → Ci / Cni = 10 → 91% forme ionisée (Log 10 = 1)
- A pH intestinal (5 < pH <7) : partie moyenne de l’intestin (surface de contact importante et forte
vascularisation)
o Les acides (pKa > 7,5) et bases faibles (pKa < 5) : absorption indépendante du pH
o Les acides (3 < pKa < 7,5) et bases (5 < pKa < 11) : absorption dépendante du pH

Acides et bases fortes fortement dissociés, peu absorbables
- Au niveau de l’estomac et début intestin ( 1-2 < pH < 5) : acides absorbables car peu dissociés (ionisation
faible)
- Au niveau de l’extrémité de l’intestin grêle et du côlon (pH = 8) : bases fortes absorbables
c) Liposolubilité – coefficient de partage Kp (Log P ou Log D)
- Kp = Rapport des concentrations de la substance à l’équilibre dans 2 solvants non mmmiiiscibles
- Kp = Concentration dans le solvant 1 non polaire / concentration dans le solvant 2 aqueux (octanol / eau)
Kp est le reflet de la liposolubilité de la forme non ionisée du médicament → log P (pH
neutre) ou Log D (fonction des pH)
0 < Log P < 4 : meilleure absorption (+ Log P augmente, + l’hydrophilie diminue)
Seule la forme liposoluble peut être absorbée par simple diffusion
- Vitesse de transfert sera fonction :
o Du Kp entre couche lipidique de la membrane et le milieu
o Du gradient de concentration de la forme liposoluble
Le caractère lipidique de la membrane facilite le passage des substances liposolubles, dont le coefficient de
partage est élevé
Log P ou Log D
Plus Cp est grand, plus l’affinité pour les lipides est grande et plus l’affinité pour les membranes et tissus à
dominance lipidique (graisse ou cerveau) est grande → effet de stockage
Ex : Thiopental et phénobarbital
Caractéristiques structurelles et physico-chimiques semblables mais Cp du Thiopental est
70 fois plus élevé que le phénobarbital → action rapide du Thiopental mais action plus
brève → inducteur d’anesthésie
d) Masse moléculaire et encombrement moléculaire
Diffusion des molécules par diffusion passive est liée à l’agitation moléculaire
Diffusion des molécules plus importante que la masse moléculaire est faible
2) Caractéristiques liées au sujet = caractéristiques du milieu
Résorption influencée :
- Par niveau de vascularisation
- Par pH des divers organes constituant le tractus digestif
- Par sécrétion digestives (Bile alcalinise le milieu et formation de complexes)
- Par transporteurs d’efflux et d’influx : affinité de la molécule pour ces transporteurs
- Résorption gastro-intestinale :
o Possible par diffusion passive
Si dissolution principe actif (hydrosolubilité) pour substance liposoluble et non ionisée
o Possible par l’intermédiaire de transporteurs
4) Facteurs limitants essentiels de la résorption
- pH et état d’ionisation
- Dissolution (étape de libération)

3 règles :
o Résorption en solution est facilitée par rapport à une forme solide
o Résorption sous forme de sel (sel de Na ou K) > acide libre
o Résorption améliorée par réduction de la taille des particules solides
- Vidange gastrique : modification de la vitesse de résorption et du temps de latence
Augmentation de la vidange gastrique → augmentation de l’absorption (estomac atteint
plus vite) et diminution du temps de séjour dans l’estomac (destruction par acidité)
- Débit sanguin intestinal : pour substances très liposolubles
- Temps de transit intestinal : Augmentation ou diminution de la motilité intestinale → augmentation ou
diminution du temps de transit → absorption
- Transporteurs (transporteurs d’efflux : MDR our MRP)
- Aliments : diminution de l’absorption car diminue les contacts avec les membranes et / ou interactions
chimiques par adsorption (tétracyclines et produits laitiers ↘50%) ou composants
- Médicaments
- Formulations pharmaceutiques
5) Paramètres pharmacocinétiques de la résorption
- Coefficient de résorption = f
C’est la fraction ou le pourcentage de dose administrée qui se trouve résorbée au niveau
de la muqueuse GI sans distinction entre la forme initiale et ses métabolites. Valeurs de f
entre 0 et 1.
- Vitesse à laquelle a lieu cette résorption
Cette vitesse conditionne le moment d’obtention du pic de concentration maximale :
Cmax et Tmax
2) Effets de premier passage
1) Définitions
Perte de médicaments par métabolisme avant son arrivée dans la circulation générale,
dès son premier contact avec l’organe responsable de sa biotransformation
- Effets de premier passage et voies d’administration :
o Voie intra-artérielle : voie de référence
o Voie intraveineuse : 1er passage pulmonaire ; en théorie, voie de référence
o Voie orale : 1er passage intestinal, hépatique, pulmonaire
o Voie rectale :
Si résorption au niveau inférieur : pas de 1er passage hépatique
Si résorption au niveau supérieur : 1er passage hépatique
o Voies IM, SC : comparables à la voie IV (intraveineuse)
2) Considérations générales
- Effet pas toujours défavorable :
o Perte de médicaments : diminution de l’effet thérapeutique
o Mais apparition de métabolites actifs : augmentation de l’effet thérapeutique
- Effet pas toujours prévisible
- Phénomène saturable et atténuation dû au fait d’une élévation de dose administrée pour saturer les
réactions enzymatiques impliquées

3) Paramètres pharmacocinétiques
Coefficient d’extraction de l’organe = E
- C’est la fraction du médicament résorbé, extrait lors du 1er passage au niveau de l’organe atteint et
soustraite à la circulation générale
- Valeur entre 0 et 1
Fraction de médicament qui a échappé aux effets de 1er passage = F’
- F’ = 1 – E
- Soit f = le coefficient de résorption
F x F’i x F’h x F’p = fraction de dose arrivant dans la circulation générale
(i : intestin h : foie p : poumon)
3) Biodisponibilité
1) Biodisponibilité absolue
Définition
- Se définit comme le pourcentage ou la fraction de substance en solution qui, après administration, atteint
la circulation générale
- Se définit par :
o La quantité de principe actif disponible au site d’action
o La vitesse avec laquelle la quantité est disponible à ce site
Le premier paramètre reflète l’intensité de l’absorption, conditionnée par 2 facteurs
- La quantité résorbée
- La quantité éliminée par les différents effets de 1er passage
F = f x F’ avec f : coefficient de résorption F’ : fraction de médicament qui a
échappé aux effets de 1er passage
Le second paramètre reflète l’aspect cinétique donc la vitesse de résorption
- Détermination de Tmax et Cmax
- Durée de résorption = TA = Tmax – temps de latence (Lag T)
2) Mode de détermination de F
En théorie, par voie IV, l’absorption est totale et immédiate → F = 1
- La biodisponibilité absolue F est évaluée :
o Par comparaison des SSC (surfaces sous la courbe) des concentrations plasmatiques
o Par comparaison des quantités éliminées dans les urines sous forme inchangée Qe
Après administration per os et après administration dans le flux sanguin (voie IV) du
principe actif
Chez un même individu et en général à une même dose
Cinétique plasmatique – D IV
SSC = AUC = aire sous la courbe
Détermination de F
 6
7
6
7
1
/
7
100%