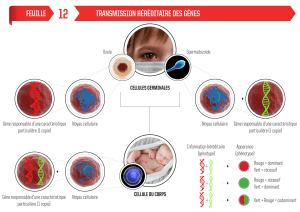
Génétique du Cancer

Génétique du Cancer
1. Facteurs génétiques du cancer révélés par
analyse de la génétique prémoléculaire
2. Oncogènes
3. Anti-oncogènes
4. Hypothèse de carcinogénèse de deux-temps à
pleusieures étapes.

Facteurs génétiques du cancer révélés par analyse
de la génétique prémoléculaire
1. Anomalies chromosomiques avec risque augmenté
du cancer.
2. Mutations monogéniques et cancer.
3. Maladies monogéniques associées avec instabilité
chromosomique et cancer.
4. Cancers de la nature polyfactorielle.

Aberrations chromosomiques constitutionnelles
avec risque du cancer
maladie anomalie type de importance
génétique chromosomique prédisposition clinique
au cancer
Syndrome de Down trisomie 21 leucémie cause la plus fréquente de
(Mongolisme) retard mental modéré; le
risque est lié à l’âge
maternel
Syndrome de 47, XXY Cancer du phénotype de type masculin
Klinefelter Sein associé à des anomalies
caractéristiques; incidence
1/1000 garçons
Syndrome de 45, X Carcinome phénotype de type féminin
Turner de l’ovaire associé à des anomalies
caratéristiques

Mutations monogénique et cancer
Maladie mode tumeur
génétique d’hérédité
Exostose multiple AD Chondrosarcome
héréditaire
Neurofibromatose AD Neurofibrosarcome
Néoplasies AD Carcinome parathyroïd
multiples Carcinome thyroïd
Polypose AD Cancer du côlon
familiale du côlon
Tylosis AD Cancer de l’oesophage
Syndrome familial AD Cancer du côlon
du cancer Cancer du sein etc
Rétinoblastome AD Rétinoblastome
AD=autosomique dominant

Neurofibromatose
1. Features cliniques majeures
La feature seule constante de cette maladie est la
presence de taches café au lait, tumeurs fibromateuses de
la peau.
(1) Von Recklinghausen neurofibromatose(NF1):
Taches café au lait.
Nombreuse neurofibromes qui dérivent du système
nerveux périphérique, augmentent en nombre et taille
avec âge.
Autre tumeurs d’origine des crêtes neurales,
particulièrement les gliomes du nerf optique et
neurofibrosarcome sont aussi présents.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
1
/
90
100%