>

20 La Lettre du Neurologue - Suppl. Les Actualités au vol. X - n° 6 - juin 2006
ACTUALITÉS
neurosciences
neurosciences
> Behavioural Brain Research
> European Journal
of Neuroscience
> Nature
> NeuroImage
> Neuron
> Molecular psychiatry
>Science
> Trends in Neuroscience
> Neurobiology
of Learning and Memory
> PNAS
>
ACTUALITÉS
neurosciences
neurosciences
E
n 1984, Glenner et Wong décou-
vraient que le peptide Aβétait le
constituant de la substance amyloïde
présente au niveau des vaisseaux ménin-
gés des patients souffrant de maladie
d’Alzheimer (MA) et de trisomie 21. Cette
découverte inaugura une succession de
travaux qui allaient bâtir l’hypothèse
amyloïdergique selon laquelle l’accumu-
lation de peptide Aβserait le primum
movens de la MA. Le peptide Aβrésulte
du clivage de la protéine précurseur de
l’amyloïde (APP), codée par le gène APP,
localisé sur le chromosome 21. Cette
localisation a donc fait attribuer la pré-
valence élevée de la maladie d’Alzhei-
mer chez les patients trisomiques 21
(> 50 % entre 50 et 60 ans) à la copie
supplémentaire du gène APP. De même,
c’est logiquement dans l’APP que furent
identifiées en 1991 les premières muta-
tions responsables des formes autoso-
miques dominantes de la MA. En 1995,
des études de linkage identifiaient des
mutations sur les gènes des préséni-
lines 1 et 2 (PSEN1/PSEN2) qui permi-
rent de mieux comprendre les voies de
maturation de l’APP. En effet, les présé-
nilines se sont avérées être des parte-
naires du complexe gamma-sécrétase qui
clive l’APP au niveau de la terminaison
transmembranaire du peptide Aβ. Un
argument principal en faveur de l’hypo-
thèse amyloïdergique est que à la fois
les mutations de l’APP et celles de
PSEN1/PSEN2 ont comme conséquence
commune une surproduction du pep-
tide Aβ, notamment de sa forme la plus
longue, Aβ42, qui possède un potentiel
d’agrégation fibrillaire beaucoup plus
important que le peptide Aβ40.
Grâce à la collaboration de plusieurs
équipes de neurologie et de neuropa-
thologie françaises, nous recherchons
les mutations des gènes APP et PSEN1-2
dans les familles atteintes de formes
autosomiques dominantes de la MA.
Nous disposions ainsi de 12 familles
dans lesquelles aucune mutation sur
APP et PSEN1-2 n’avait été identifiée.
Nous avons donc décidé de rechercher
dans ces familles une modification du
dosage génique de l’APP en utilisant une
méthode semi-quantitative fondée sur
une PCR multiplex de courts fragments
fluorescents (QMPSF). Cette méthode a
permis d’identifier cinq duplications du
locus APP de taille différente, variant
de 0,58 à 6,7 Mb, et contenant de 5 à
12 gènes. Dans chacune des 5 familles,
la duplication était retrouvée chez
chaque patient et était absente chez
les apparentés indemnes âgés de plus
de 60 ans. Aucun des patients ne pré-
sentait de retard mental, de signes dys-
morphiques ou de pathologies classi-
quement associées à la trisomie 21. Le
phénotype dans ces familles était carac-
térisé par une angiopathie amyloïde
cérébrale (AAC) sévère. La démence
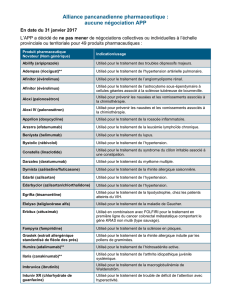
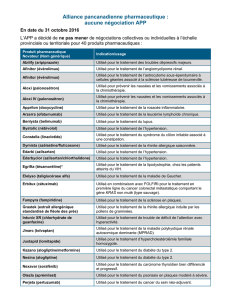
Duplication du gène de l’APP
et maladie d’Alzheimer
D. Hannequin*, D. Campion*
>
Rovelet-Lecrux A, Hannequin D, Raux G et al. APP locus duplication causes autosomal dominant
early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 2006;38(1):24-6.
>
* Inserm U614, faculté de médecine, et CMRR
de Rouen.

La Lettre du Neurologue - Suppl. Les Actualités au vol. X - n° 6 - juin 2006 21
de l’APP suffisant à causer une forme
précoce de MA, il paraît licite de sug-
gérer qu’une augmentation plus limitée
de l’expression de l’APP, par exemple
par le biais de mécanismes régulateurs,
puisse causer ou favoriser une forme
tardive de la maladie.
Ces résultats valident l’imputabilité de
la copie supplémentaire d’APP dans la
MA associée à la trisomie 21, car la plus
petite duplication ne concerne que
4 gènes en dehors de l’APP. La preuve
absolue serait apportée par la décou-
verte d’une MA causée par duplication
limitée au seul gène APP. Le phénotype
de nos observations, caractérisé par la
fréquence des hématomes, devrait être
proche de celui affectant les patients
trisomiques 21 âgés de plus de 50 ans.
En fait, les hématomes intracérébraux
causés par l’angiopathie amyloïde sont
rarement rapportés chez les patients tri-
somiques 21 atteints de MA. Une espé-
rance de vie moyenne estimée actuelle-
ment à 60 ans et des causes de décès
non liées à la démence pourraient expli-
quer cette différence de prévalence. Il
paraît donc nécessaire de collecter une
série plus importante de patients por-
teurs de duplications de l’APP pour
déterminer la possible variabilité de la
sévérité de l’AAC et la fréquence des
hématomes intracérébraux.
Une AAC sévère s’exprimant essentielle-
ment par des hématomes, parfois par
des accidents ischémiques, et associée
à des anomalies de la substance blanche
ou à des microbleeds en IRM est connue
débutait entre 42 et 59 ans. Dans
4 familles, alors que la majorité des
patients répondait aux critères de MA
probable, un patient au moins présen-
tait une démence vasculaire au décours
d’un hématome intracérébral. Les exa-
mens anatomopathologiques de 5 cer-
veaux appartenant à trois familles (dont
celle indemne d’hématome) ont révélé
une AAC sévère, des dépôts amyloïdes
diffus et sous forme de plaques ainsi
que des dégénérescences neurofibril-
laires correspondant à un stade V-VI de
Braak. L’angiopathie était extensive
dans toutes les régions, touchait les
vaisseaux leptoméningés, les artérioles
superficielles et intraparenchymateuses,
les capillaires et veinules. Les immuno-
marquages ont montré que l’AAC était
majoritairement composée de peptide
Aβ40 alors que les plaques avaient un
centre marquant fortement Aβ42, entouré
d’un marquage Aβ40.
Ce travail fournit un argument majeur à
l’hypothèse amyloïdergique, puisqu’il
démontre qu’une augmentation de
l’expression de l’APP suffit à déclencher
la MA. Il s’agit d’une nouvelle illustra-
tion montrant qu’un mécanisme purement
quantitatif peut causer une maladie
neurodégénérative, au même titre que
des duplications ou des triplications du
gène de l’α-synucléine sont responsables
de certaines formes autosomiques
dominantes précoces de la maladie de
Parkinson. Ce type de mécanisme, mais
de moindre ampleur, pourrait également
s’appliquer aux formes communes de
MA. En effet, une copie supplémentaire
dans de rares cas de mutations ponc-
tuelles de l’APP, au niveau de la région
codante du peptide Aβ. Dans la plupart
des cas, cette AAC coexiste avec les
lésions caractéristiques de MA (plaques
séniles et dégénérescences neurofibril-
laires), comme dans les présentes obser-
vations causées par des duplications de
l’APP. En revanche, des mutations aux
codons APP 692-693 sont responsables
d’une AAC associée à des dépôts intra-
parenchymateux de peptide Aβ, mais sans
dégénérescence neurofibrillaire, et la
mutation APP L705V cause une démence
due à la seule AAC sans dépôt paren-
chymateux. Il reste donc à comprendre
les mécanismes selon lesquels les pep-
tides Aβ42 et Aβ40 se déposent préfé-
rentiellement au niveau du parenchyme
cérébral ou des parois vasculaires, avec
une palette d’expression de l’AAC pou-
vant être sévère et isolée, sévère et
associée à la MA, comme dans nos pré-
sentes observations, ou limitée comme
celle habituellement observée dans les
formes communes de MA.
Enfin, soulignons combien la description
de telles observations dépend des inter-
actions entre spécialistes de la patho-
logie vasculaire et des démences. Ainsi,
en pratique clinique, chez un patient de
moins de 60 ans atteint d’un hématome
intracérébral spontané, l’enquête fami-
liale doit rechercher à la fois des cas
similaires et une pathologie démentielle
répondant aux critères de MA. De même,
chez un patient atteint de MA précoce,
l’existence d’anomalies de la substance
blanche ou d’AVC chez un apparenté incite
à rechercher une duplication de l’APP.
■
1
/
2
100%