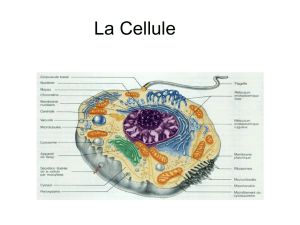
A. Rôle des ADN polymérases TLS sur les ADN non-B

T
TH
HÈ
ÈS
SE
E
En vue de l'obtention du
D
DO
OC
CT
TO
OR
RA
AT
T
D
DE
E
L
L’
’U
UN
NI
IV
VE
ER
RS
SI
IT
TÉ
É
D
DE
E
T
TO
OU
UL
LO
OU
US
SE
E
Délivré par l'Université Toulouse III - Paul Sabatier
Discipline ou spécialité : Cancérologie
JURY
Agnès CORDONNIER Chargé de Recherche, ESBS, Illkirch Rapporteur
Filippo ROSSELLI Directeur de Recherche, IGR, Villejuif Rapporteur
Philippe PASERO Directeur de Recherche, IGH, Montpellier Examinateur
Bernard DUCOMMUN Professeur de l'Université de Toulouse Président
Jean-Sébastien HOFFMANN Directeur de Recherche, IPBS, Toulouse Directeur de thèse
Ecole doctorale : Biologie-Santé-Biotechnologies de Toulouse
Unité de recherche : Institut de Pharmacologie et de Biologie Structurale
,
Equipe "Instabilité Génétique et cancer", UMR5089 CNRS/UPS
Directeur(s) de Thèse : Jean-Sébastien Hoffmann
Co-directrice de Thèse : Nadine Puget
Rapporteurs : Agnès Cordonnier, Filippo Rosselli
Présentée et soutenue par
Laurie
REY
Le 19 Juin 2009
Titre :
L'ADN Polymérase eta humaine est requise pour la stabilité des séquences particulières de
l'ADN en absence de stress exogène : rôle dans la réplication et/ou dans la réparation par
recombinaison homologue ?


1
Je voudrais tout d’abord remercier le Dr. Agnès Cordonnier et le Dr. Filippo Rosselli
pour avoir accepté de juger mon travail de thèse. Je voudrais aussi remercier sincèrement le
Dr. Philippe Pasero et le Pr. Bernard Ducommun pour avoir accepté de siéger à mon jury de
thèse. Je remercie particulièrement Philippe pour ses conseils (et ceux d’Hélène Tourrière) au
niveau des expériences de ChIP. Je vous remercie pour les discussions enrichissantes que
nous avons pu avoir lors de ma soutenance de thèse.
Je voudrais ensuite remercier les personnes qui se sont succédées à la direction de ma
thèse. Tout d’abord, Nadine merci de m’avoir fait confiance, d’avoir initié ce projet et de
m’avoir suivi au cours de la première année. Christophe et Jean-Sébastien, merci de m’avoir
laissé autant d’autonomie, de m’avoir considérée plus comme un chercheur que comme une
étudiante et surtout merci de m’avoir donné l’opportunité de partir en congrès international
toute seule. C’était pour moi une expérience très riche et qui m’a beaucoup apporté aussi bien
pour la suite de ma thèse que pour la suite de ma carrière.
Un grand merci à toute l’équipe IGC : merci Marie et Anne pour avoir répondu à
toutes mes questions scientifiques et de vie pratique du laboratoire, merci Valérie de
reprendre le sujet et de faire vivre ce que je n’ai pas eu le temps de finir, merci Eddy de
m’avoir supportée dans le bureau avec mes moments de détresse face à un ordinateur parfois
incompréhensible, merci Anne de redonner un peu de vie au labo que j’ai (un peu) déserté
pendant la rédaction. Merci Fanny, on a su se serrer les coudes aussi bien pour les manips,
pour les coups durs, mais aussi lors de la rédaction… Bonne chance pour la suite !
Je voudrais aussi dire un grand merci aux personnes qui ont quitté l’équipe : Dennis,
François et Yvan pour avoir continué à m’aider et à me soutenir dans les moments difficiles.
Un grand merci à Oriane : je suis déçue que le « destin » nous ait séparées, j’aurais vraiment
aimé travailler avec toi, cela aurait été très stimulant. Mais je suis sûre que l’avenir va te
sourire car tu le mérites vraiment ! Continues de te battre et surtout restes positive.
Cette thèse m’a amenée à travailler dans le laboratoire dirigé par Martine Yerle à
l’INRA d’Auzeville. Je voudrais particulièrement remercier Martine pour m’avoir autorisée à
faire des manips dans son laboratoire et à Florence Mompart pour son aide et tous ses
conseils. J’ai réussi à mettre en place la manip de FISH à l’IPBS mais sans vos compétences
la tâche aurait été beaucoup plus difficile. Par la suite, j’ai aussi eu la chance de travailler en
collaboration avec Julia Sidorova et Raymond Monnat de l’université de Washington à
Seattle. Les conseils inépuisables de Julia aussi bien techniques que scientifiques et l’aide de

2
Raymond Monnat en terme de réflexion sur l’article m’ont permis d’envisager mes résultats
sous un autre angle. Je vous en suis très reconnaissante. Je remercie aussi Denis Biard,
Patricia Kannouche et Karen Vasquez pour leur contribution au cours de ma thèse.
Je voudrais aussi dire un grand merci à mes amis : Amandine, Céline et Claire pour
m’avoir sorti de ma thèse de temps en temps, pour m’avoir permis de passer de bonnes
soirées, pour m’avoir remonté le moral quand ça n’allait pas et tout simplement pour votre
présence. Merci aussi à Aline, Fanny, Marielle et Sébastien pour avoir supporté mes plaintes !
Un merci tout spécial à Claude pour tes innombrables conseils, pour ton aide dans mes
moments de doute au cours de ma thèse, pour ton écoute et ton soutien, mais aussi merci de
croire en moi.
Un merci tout particulier à François : sans toi, je ne sais pas si je serais encore dans le
monde de la recherche, sans ta passion communicative pour la science, je ne sais pas si
j’aurais pu continuer. Merci d’avoir rempli ma vie de bonheur, de m’avoir ouvert les yeux sur
tant de choses, de m’avoir permis de retrouver un tant soit peu ma confiance en moi et mon
équilibre, de m’avoir redonner envie de me battre pour ce que je crois juste et utile et merci
tout simplement pour ta présence au quotidien à mes côtés et pour m’avoir permis de
retrouver le calme de mon enfance à la campagne.
Je voudrais terminer en disant un grand merci à ma famille : tout particulièrement à
mes parents, Marie-Claude et Christian, sans qui je n’aurais jamais pu arriver à réaliser mon
rêve et à mon frère, Frédéric. Merci de m’avoir fait confiance, de m’avoir toujours laissée
faire mes propres choix sans parfois vraiment comprendre pourquoi, d’avoir cru en moi et tout
simplement pour votre amour et votre soutien au quotidien. Merci à mes grands parents
Paulette et René, pour votre amour et pour m’avoir permis de trouver toujours une porte
ouverte quand j’avais besoin de me ressourcer. J’ai enfin une pensée toute particulière pour
mes grands parents, Eliette et Edmond, et mon oncle, Benoit, qui sont décédés au cours de ma
thèse et je les remercie du fond du cœur de m’avoir montré à quel point le courage était
important dans la vie ! J’espère un jour arriver à comprendre pourquoi une maladie comme le
cancer peut être si injuste et si terrible et surtout comment chaque patient qui souffre peut être
soigné efficacement. J’espère que l’avenir me permettra d’y consacrer au moins une partie de
ma vie.

3
TABLE DES
MATIERES
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
1
/
268
100%