gènes suppresseurs de tumeurs

N°7 Juillet 2013
ONCO
news
la lettre CAHPP sur la cancérologie
Un regard différent
L
e
s
t
h
é
r
a
p
i
e
s
c
i
b
l
é
e
s
T
u
m
e
u
r
s
s
o
l
i
d
e
s
e
t
l
i
q
u
i
d
e
s

2
Bonjour,
La CAHPP s’est volontairement concentrée sur l’oncologie ces dernières années et ONCO
NEWS en est l’illustration.
Parmi les secteurs difciles dans lesquels nous avons décidé de nous investir, la cancérolo-
gie, l’addiction, la cardiologie, la dialyse sont autant de dossiers sur lesquels nous portons
nos efforts avec le médico-social.
Il ne s’agit pas que de diffuser des études, analyses ou références que suscite l’investisse-
ment de tout ceux qui ont à combattre les effets de la maladie mais d’apporter une réexion
permanente, regrouper les informations et démontrer la qualité de la prise en charge que
toutes les équipes, scientiques et personnels médicaux et paramédicaux apportent à ce com-
bat permanent.
Nous avons toujours considéré que, certes, la négociation à laquelle s’engage chaque année
la CAHPP est importante mais combien est aussi important l’environnement mais aussi com-
bien il est indispensable de prendre quelques risques et de consacrer des moyens à l’infor-
mation, la communication.
Nous espérons que cette revue, souvent réclamée dans les milieux professionnels, vous ap-
portent une nouvelle fois un plus et nous vous souhaitons de bonnes vacances… la rentrée
sera rude…
Yvon BERTEL VENEZIA
Président Directeur Général CAHPP
Directeur de la publication Onconews
Editorial

3
Sommaire
N°7 Juillet 2013
ONCO
news
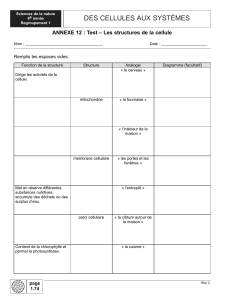
1 Définitions ........................................................................................................... 6
A. Cellules saines/cellules cancéreuses ........................................................................................... 6
B. Oncogènes/gènes suppresseurs de tumeur .................................................................................. 7
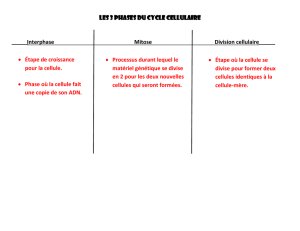
C. Le cycle cellulaire ..................................................................................................................... 9
D. Les thérapies ciblées : c’est quoi ? ............................................................................................ 10
2 Les thérapies ciblant la cellule tumorale au niveau membranaire ........................11
A. La famille HER ......................................................................................................................... 12
1. Description
2. Mécanisme d’action
3. Dans quelles tumeurs ?
4. Les molécules
5. Cas des ITK irréversibles
6. Le TDM-1
B. Kit et PDGF-R ........................................................................................................................... 16
1. La protéine Kit
a. Description
b. Mécanisme d’action
2. PDGF-R
a. Description
b. Mécanisme d’action
3. Dans quelles tumeurs ?
4. Les molécules
5. Le chromosome de Philadelphie
C. IGF1-R ..................................................................................................................................... 18
1. Description
2. Mécanisme d’action
3. Dans quelles tumeurs ?
4. Les molécules
D. HGF et son récepteur MET ....................................................................................................... 20
1. Description
2. Mécanisme d’action
3. Dans quelles tumeurs ?
4. Les molécules

3 Les thérapies ciblant la cellule tumorale au niveau du cytoplasme .................... 22
A. La voie PI3K/AKT/mTOR .......................................................................................................... 22
1. Description
2. Mécanisme d’action
3. Dans quelles tumeurs ?
4. Les molécules
B. La voie RAF/MEK/ERK .............................................................................................................. 24
1. Description et mécanisme d’action
2. Dans quelles tumeurs ?
3. Les molécules
C. La famille SKF .......................................................................................................................... 26
1. Description.
2 Mécanisme d’action
3. Dans quelles tumeurs ?
4. Les molécules
D. Les inhibiteurs du protéasome ................................................................................................. 26
1. Description
2. Mécanisme d’action
3. Les molécules
4 Les thérapies ciblées agissant au niveau du cycle cellulaire ............................... 28
A. Les cyclines .............................................................................................................................. 28
1. Description
2. Mécanisme d’action
3. Les molécules
B. Les kinases Aurora .................................................................................................................... 30
1. Description
2. Mécanisme d’action
3. Les molécules
4

N°7 Juillet 2013
ONCO
news
5 Les thérapies agissant au niveau de l’environnement tumoral ............................ 31
A. Définition du stroma ................................................................................................................ 31
B. Les antiangiogèniques .............................................................................................................. 31
1. Définition de l’angiogenèse
2. La famille VEGF / VEGF-R
a. Description
b. Mécanisme d’action
c. Les molécules
. Les molécules agissant au niveau du ligand
. Les molécules agissant au niveau du recepteur
3. Les autres inhibiteurs de l’angiogenèse
a. HIF-1
b. Les agents antivasculaires
. Les agents ciblant la tubuline
. Les flavanoides
C. Les inhibiteurs des métalloprotéinases ...................................................................................... 35
1. Mécanisme d’action
2. Description
3. Dans quelles tumeurs ?
4. Les molécules
6 Les anticorps monoclaux ................................................................................... 37
A. Structure des anticorps monoclonaux ...................................................................................... 37
B. Mécanisme d’action ................................................................................................................. 38
1. Action par blocage de l’antigène ou du récepteur spécifique
2. Activation des mécanismes de défense de l’hôte
a. Cytotoxicité cellulaire dépendante des anticorps ADCC
b. Cytotoxicité dépendante du complément CDC
C. Nouvelles approches de l’utilisation des anticorps monoclaux en cancérologie ....................... 40
1. Les anticorps conjugués
2. Les anticorps bi-spécifiques
D. Les anticorps monoclonaux utilisés en cancérologie ................................................................ 40
7 Conclusion ........................................................................................................ 41
8 Bibliographie ..................................................................................................... 42
5
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
1
/
44
100%