Rapport de stage 2006 (pdf de 450ko)

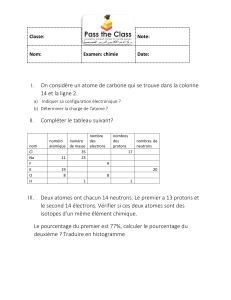
1
SENILLE Violette
M1 BBSG - Luminy
Stage en laboratoire
Structure d’un mutant
d’une toxine de scorpion
∆
1-7
AOSK1
Responsable : Hervé DARBON - Encadrant : Stéphane GELY
Architecture et Fonction des Macromolécules Biologique
CNRS – 31, chem. Joseph Aiguier
13420 Marseille Cedex 20

2
I. Introduction.
La protéine OSK1 est une toxine isolée du venin d’un scorpion asiatique, et qui
a pour cible les canaux ioniques spécifiquement perméables au potassium.
Des analogues structuraux d’OSK1 on été fabriqués. Les tests
pharmacologiques de ces mutants ont montré que certains seraient de meilleurs
inhibiteurs que la toxine naturelle, sur certains types de canaux ioniques.
Le but de ce stage est de résoudre la structure d’un de ces mutants (∆
1-
7
AOSK1) par RMN bidimensionnelle du proton, pour tenter de déterminer les
différences structurales à l’origine du changement de spécificité observé.
A. OSK1.
OSK1 est un inhibiteur des canaux potassium de la famille Kv1, activés par
des variations de potentiel de membrane (Kv1.1, Kv1.2 et Kv1.3). Il agit
également sur des canaux potassium activés par le calcium, mais de façon moins
importante.
Cette protéine bloque notamment deux canaux impliqués dans l’activation des
Lymphocytes T et B chez l’homme, elle a été étudiée dans le but de créer des
peptides immunosuppresseurs.
OSK1 fait partie de la famille α-KTx3 (kaliotoxines, agitoxines). C’est une
protéine de 38 amino-acides, dont la structure a été résolue par RMN (Mouhat et
al., 2004b).
Elle est composée d’une hélice α
connectée à un feuillet
β antiparallèle. La partie N-terminale
(résidus 2 à 6) constitue le 3
ème
brin
du feuillet β.
La structure est stabilisée par trois
ponts disulfures : 2 ponts connectent
l’hélice au feuillet, le 3
ème
pont
connecte le feuillet à la boucle, qui
sépare le feuillet de l’hélice.
L’étude d’OSK1 a démontré qu’elle interagit avec les canaux par
l’intermédiaire de son feuillet β. Deux résidus sont notamment impliqués dans
cette interaction : la Lysine 27 et la Phénylalanine 25.

3
B. AOSK.
AOSK est un analogue structural d’OSK1 : deux mutations ont été introduites
au niveau de l’hélice (Lys16 et Asp20), modifiant localement la charge de la
molécule et augmentant considérablement la réactivité d’AOSK1.
En effet, AOSK1 est 5 fois plus active qu’OSK1 sur les canaux Potassium, mais
ne présente pas de changements significatifs avec les canaux Calcium
dépendants
Pour trouver l’origine de ces propriétés, plusieurs mutants d’AOSK1 ont été
fabriqués : les extrémités ont été raccourcies jusqu’aux Cystéines
(indispensables pour obtenir un repliement correct).
(Mouhat et al., 2004b)
C.
∆
∆∆
∆
1-7
AOSK1.
Le mutant étudié au cours de ce stage est ∆
1-7
AOSK1 : il possède les deux
mutations dans l’hélice (AOSK1) et le 1
er
brin du feuillet β a été tronqué dans sa
totalité (∆
1-7
).

4
Une étude pharmacologique a été menée sur OSK1, AOSK1 et les différents
mutants. Les modifications introduites chez ∆
1-7
AOSK1 engendrent plusieurs
changements d’activité :
- il est plus sélectif avec les canaux de type Kv1.1 et Kv1.3, mais moins actif.
- il perd toute activité sur Kv1.2 et les canaux Ca dépendant.
(Mouhat et al., 2004b)
∆
1-7
AOSK1 possède donc une plus grande sélectivité qu’OSK1 sur certains
canaux Potassium. Mais ce mutant est beaucoup moins actif qu’OSK1. Les deux
mutations et la suppression de la partie N-terminale de la toxine seraient donc à
l’origine de ces changements.
Pour tenter d’expliquer ce phénomène, il est nécessaire de réaliser un
arrimage moléculaire entre ∆
1-7
AOSK1 et les différents types de canaux.
Durant ce stage, nous ne ferons que la première étape, c’est-à-dire la
résolution de la structure 3D de ∆
1-7
AOSK1.

5
II. Matériel et méthodes.
Pour déterminer la structure de ∆
1-7
AOSK1, nous allons utiliser la Résonance
Magnétique Nucléaire (RMN) homonucléaire (qui n’utilise que les protons).
A. Les spectres RMN.
Tous les protons d’une protéine auront des fréquences de résonance
différentes. En effet, la protéine possède des champs magnétiques locaux qui
vont provoquer de petits changements de leur fréquence.
Or la résonance d’un proton va influencer la résonance d’un autre proton si
ceux-ci sont couplés dans la protéine :c’est-a-dire s’il sont proches (separes par
trois liaisons au plus (couplage scalaire) ou 5 A au plus (couplage dipolaire).
Cette influence peut se voir sur différents spectres RMN : ils se trouvent sous
forme de pics de corrélation ayant pour coordonnées les fréquences des deux
protons.
Plus le couplage entre deux protons est fort (lorsqu’ils sont proches par
exemple), plus l’intensité du pic correspondant sera forte.
Sur le spectre COSY apparaissent les couplage entre des protons, qui sont
reliés par moins de 3 liaisons covalentes (comme les protons dans un acide
aminé) : c’est le couplage scalaire.
Le spectre TOCSY fait apparaître des pics supplémentaires, entre des protons
séparés par plus de 3 liaisons covalentes : c’est le couplage relayé. Un proton
supplémentaire fera office de relais entre les deux protons couplés.
 6
7
8
9
10
11
6
7
8
9
10
11
1
/
11
100%