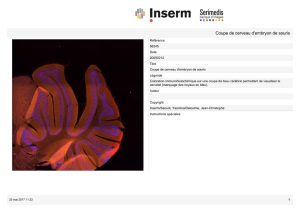
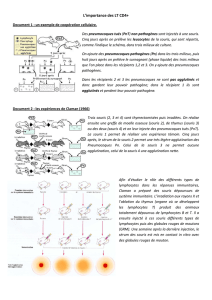
Étude de l`impact de la grossesse sur l`hépatite auto

Université de Montréal
Étude de l’impact de la grossesse sur l’hépatite
auto-immune dans un modèle expérimental murin
par
Sara Bourbonnais
Département de Microbiologie et Immunologie
Faculté de Médecine
Mémoire présenté à la Faculté de Médecine
en vue de l’obtention du grade de M.Sc
en Microbiologie et Immunologie
Septembre 2013
© Sara Bourbonnais, 2013

ii
Université de Montréal
Faculté des études supérieures et postdoctorales
Ce mémoire intitulé :
Étude de l’impact de la grossesse sur l’hépatite auto-immune
dans un modèle expérimental murin
Présenté par :
Sara Bourbonnais
A été évalué par un jury composé par :
Dre Hélène Decaluwe, présidente-rapporteuse
Dr Fernando Alvarez, directeur de recherche
Dre Valérie Abadie, membre du jury

iii
Résumé
L’hépatite auto-immune (HAI) est une maladie chronique caractérisée par une destruction
progressive du parenchyme hépatique par le système immunitaire. La majorité des patients
atteints d’HAI sont des femmes (75% à 90% des cas). L’amélioration des traitements au cours
des dernières années a permis à un grand nombre de ces femmes de devenir enceintes. Pendant
la grossesse, une rémission spontanée de la maladie a pu être observée chez les femmes
atteintes d’HAI. Cette rémission est temporaire et elle est généralement suivie d’une rechute
suite à l’accouchement (post-partum). Les causes exactes de cette rémission associée à la
grossesse et de la rechute post-partum ne sont pas connues à ce jour. Nous avons donc tenté de
reproduire ces phénomènes dans un modèle murin d’HAI développé dans notre laboratoire,
afin de déterminer les mécanismes possiblement impliqués.
Notre modèle d’HAI consiste en une xéno-immunisation de souris C57BL/6 avec les auto-
antigènes impliqués dans l’HAI de type 2 chez l’humain. Nous avons ainsi accouplées des
souris préalablement xéno-immunisées, puis nous les avons sacrifiées au début de la 3e
semaine de gestation ou 2 à 3 semaines post-partum, afin d’évaluer les dommages hépatiques
et afin d’étudier la réponse immunitaire. Comme chez les femmes atteintes d’HAI, les souris
présentent une rémission de la maladie pendant la grossesse. Nous en sommes venus à cette
conclusion par l’observation d’une diminution de l’inflammation hépatique, des niveaux de
transaminases sériques et des titres d’auto-anticorps circulants. À l’inverse des humains, les
souris xéno-immunisées ne présentent pas de rechute post-partum. Une analyse des cellules
régulatrices (cellules T régulatrices et cellules B productrices d'IL-10) suggère une implication
des Tregs hépatiques dans la rémission, car ceux-ci sont augmentés pendant la gestation. Ces
Tregs hépatiques sont majoritairement d’origine thymique et ne semblent pas particulièrement
attirés au foie en réponse à l’inflammation. La polarisation TH2 est un phénomène connu
pendant la grossesse, par contre elle ne semble pas influencer la réponse auto-immune dans
nos souris. Une meilleure compréhension des mécanismes d’immunosuppression observés lors
de la grossesse pourrait mener au développement d’une thérapie mieux ciblée.
Mots-clés : Auto-immunité, tolérance immunitaire, hépatite auto-immune, foie, grossesse,
gestation, post-partum, fœtus, hormones, lymphocytes T régulateurs.

iv
Abstract
Autoimmune hepatitis (AIH) is a disease of unknown aetiology, characterized by a
progressive destruction of the hepatic parenchyma by the immune system. Women are
predominantly affected by AIH, with a prevalence of 75% in type 1 AIH and 90% in type 2.
Improvement of treatments to control liver inflammation has contributed to increase the
number of pregnant patients. During pregnancy, a spontaneous, but temporary, remission of
the disease has been observed in women with AIH, often followed by post-partum relapse.
Currently, this phenomenon is not fully understood. Thus, we aim to study the mechanisms
responsible for the pregnancy-related remission and post-partum relapses in a murine model of
AIH developed in our laboratory.
Xenoimmunization of C57BL/6 mice with the antigens recognized by autoreactive cells of
type 2 AIH patients results in a loss of tolerance, with clinical and biochemical features of
AIH similar to those observed in patients. For this study, xenoimmunized mice were mated
and sacrificed at the beginning of the 3rd week of gestation or 2-3 weeks post-partum, and
liver disease was evaluated at that time as well as the immune response. As it occurs in women
with AIH, pregnancy induces a remission of the disease in our murin model. Pregnant mice
show a decrease of liver inflammation, ALT levels and circulating autoantibodies. The mice
did not show any sign of relapse during the post-partum period. Our analysis of cellular
populations with regulatory properties (regulatory T cells and IL-10 secreting B cells) revealed
an increase of hepatic Tregs during pregnancy, which suggests an implication of those cells in
the remission. Those Tregs infiltrating the liver are mainly originated from the thymus and do
not seem to be recruited to the liver in response to inflammation. Although a switch towards a
TH2 response is known to occur during pregnancy, this phenomenon does not seem to be
implicated in the remission of AIH in our murine model. A better understanding of the natural
immunosuppression occurring during pregnancy could bring important knowledge to design
new and improved therapy for AIH.
Keywords : Autoimmunity, immune tolerance, autoimmune hepatitis, liver, pregnancy, post-
partum, fetus, hormones, regulatory T cells

v
Table des matières
CHAPITRE 1 : INTRODUCTION ....................................................................................................... 1
1.1 LE SYSTÈME IMMUNITAIRE ............................................................................................................. 1
1.1.1 Le système immunitaire inné .................................................................................................. 1
1.1.2 Le système immunitaire adaptatif ........................................................................................... 2
1.2 LA TOLÉRANCE IMMUNITAIRE ........................................................................................................ 5
1.2.1 Tolérance centrale ................................................................................................................... 5
1.2.2 Tolérance périphérique ............................................................................................................ 6
1.3 L’AUTO-IMMUNITÉ ......................................................................................................................... 9
1.4 LE FOIE .......................................................................................................................................... 10
1.5 L’HÉPATITE AUTO-IMMUNE (HAI) ............................................................................................... 12
1.5.1 Historique .............................................................................................................................. 12
1.5.2 Hépatite auto-immune de type I ............................................................................................ 12
1.5.3 Hépatite auto-immune de type II ........................................................................................... 13
1.5.4 Auto-anticorps communs aux deux types d’HAI. ................................................................. 13
1.5.5 Manifestations cliniques et diagnostic .................................................................................. 14
1.5.6 Pathogénèse ........................................................................................................................... 18
1.5.7 Facteurs de prédispositions ................................................................................................... 18
1.5.8 Traitements ............................................................................................................................ 23
1.6 LES MODÈLES ANIMAUX D’HÉPATITE AUTO-IMMUNE .................................................................. 25
1.6.1 Modèle S-100 ........................................................................................................................ 25
1.6.2 Modèle de dommage hépatique induit par la Concanavaline A ........................................... 26
1.6.3 Souris SAP-IFN-γ ................................................................................................................. 27
1.6.4 Souris invalidée pour le TGF-β (TGF-β-/-) ........................................................................... 27
1.6.5 Souris Alb/GP33 ................................................................................................................... 28
1.6.6 Souris exprimant un ligand spécifique du CD28 (scFv) ....................................................... 28
1.6.7 Souris TTR-NP ...................................................................................................................... 29
1.6.8 Modèle par xéno-immunisation avec le plasmide pCMV-CTLA4-CYP2D6-FTCD ........... 30
1.6.9 Modèles basés sur l’utilisation d’un vecteur adénovirus ...................................................... 32
1.7 LE SYSTÈME IMMUNITAIRE PENDANT LA GROSSESSE ................................................................... 33
1.7.1 Modulation de la réponse immunitaire lors de la grossesse .................................................. 33
1.7.2 Mécanismes de tolérance envers le fœtus ............................................................................. 34
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
1
/
121
100%