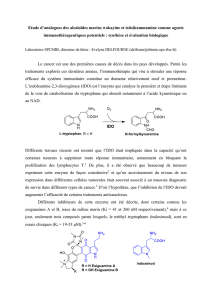
Les liquides ioniques, leur utilisation et leur role comme solvants de

Les liquides ioniques, leur utilisation et leur role comme
solvants de r´eaction catalytique
Thibaut Gutel
To cite this version:
Thibaut Gutel. Les liquides ioniques, leur utilisation et leur role comme solvants de r´eaction
catalytique. Catalyse. Universit´e Claude Bernard - Lyon I, 2007. Fran¸cais. <tel-00182860>
HAL Id: tel-00182860
https://tel.archives-ouvertes.fr/tel-00182860
Submitted on 29 Oct 2007
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-
entific research documents, whether they are pub-
lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destin´ee au d´epˆot et `a la diffusion de documents
scientifiques de niveau recherche, publi´es ou non,
´emanant des ´etablissements d’enseignement et de
recherche fran¸cais ou ´etrangers, des laboratoires
publics ou priv´es.

N° d’ordre : 173 - 2007 Année 2007
THESE
présentée
devant l’UNIVERSITE CLAUDE BERNARD - LYON 1
ECOLE DOCTORALE DE CHIMIE, PROCEDES, ENVIRONNEMENT
Pour l’obtention
du DIPLOME DE DOCTORAT
Spécialité CHIMIE
(arrêté du 25 avril 2002)
présentée et soutenue publiquement le 12 octobre 2007
par
Thibaut Gutel
Ingénieur ESCOM
LES LIQUIDES IONIQUES, LEUR UTILISATION ET LEUR ROLE
COMME SOLVANTS DE REACTION CATALYTIQUE
Directeurs de thèse :
Catherine SANTINI et Yves CHAUVIN
Jury : M. BASSET, Président
M. COLE-HAMILTON, Rapporteur
M. SIMONATO, Rapporteur
M. CHAUDRET
M. LEMAIRE
Mme OLIVIER-BOURBIGOU

2
UNIVERSITE CLAUDE BERNARD – LYON I
Président de l’Université M. le Professeur L.COLLET
Vice-Président du Conseil Scientifique M. le Professeur J.F. MORNEX
Vice-Président du Conseil d’Administration M. le Professeur J. LIETO
Vice-Président du Conseil des Etudes et M. le Professeur D. SIMON
de la Vie Universitaire
Secrétaire Général M. G. GAY
SECTEUR SANTE
Composantes :
UFR Médecine Lyon R.T.H Laënnec Directeur : M. le Professeur D.VITAL-DURANI
UFR Médecine Lyon Grange-Blanche Directeur : M . le Professeur X. MARTIN
UFR Médecine Lyon-Nord Directeur : M. le Professeur F. MAUGUIERE
UFR Médecine Lyon-Sud Directeur : M. le Professeur F.N. GILLY
UFR d’Odontologie Directeur : M. O. ROBIN
Institut des Sciences Pharmaceutiques Directeur : M. le Professeur F.LOCHER
et Biologiques
Institut Techniques de Réadaptation Directeur : M. le Professeur MATILLON
Département de Formation et Centre Directeur : M. le Professeur P.FARGE
de Recherche en Biologie Humaine
SECTEUR SCIENCES
Composantes :
UFR de Physique Directeur : M. le Professeur A.HOAREAU
UFR de Biologie Directeur : M. le Professeur H.PINON
UFR de Mécanique Directeur : M. le Professeur H. BEN HADID
UFR de Génie Electrique et des Procédés Directeur : M. le Professeur A.BRIGUET
UFR Sciences de la Terre Directeur : M. le Professeur P.HANTZPERGUE
UFR de Mathématiques Directeur : M. le Professeur M.CHAMARIE
UFR d’Informatique Directeur : M. le Professeur M. EGEA
UFR de Chimie Biochimie Directeur : M. le Professeur H. PARROT
UFR STAPS Directeur : M. le Professeur R.MASSARELLI
Observatoire de Lyon Directeur : M. le Professeur R.BACON
Institut des Sciences et des Techniques Directeur : M. le Professeur J. LIETO
de l’ingénieur de Lyon
IUT A Directeur : M. le Professeur M.C. COULET
IUT B Directeur : M. le Professeur R.LAMARTINE
Institut de Science Financière et Directeur : M. le Professeur J.C AUGROS
d’Assurance

3
A mon grand-père,

4
Les travaux exposés dans ce mémoire ont été réalisés entre septembre 2004 et octobre
2007 au Laboratoire de Chimie, Catalyse, Polymérisation et Procédés dans l’équipe de
Chimie OrganoMétallique de Surface, unité mixte CNRS-CPE Lyon. Je suis reconnaissant
envers Monsieur Gérard Pignault, directeur de CPE Lyon, pour m’avoir accueilli dans ses
locaux.
Mes remerciements vont tout d’abord à Monsieur Jean-Marie Basset, Directeur de
Recherche au CNRS, qui a bien voulu m’accueillir au sein de son laboratoire.
J’adresse mes plus vifs remerciements à Monsieur Yves Chauvin, Directeur de
Recherche honoraire à l’Institut Français du Pétrole, agissant en qualité de co-directeur. En
dépit de son éloignement, il a fait preuve d’une grande disponibilité et a été une source
constante de conseils et de réflexions. Je lui suis très reconnaissant de sa contribution au
développement de ce projet et de m’avoir fait profiter de ses connaissances et de sa grande
expérience en chimie.
Je tiens à remercier Madame Catherine Santini, Directeur de Recherche au CNRS, qui
a encadré mes travaux de thèse. Je la remercie tout particulièrement pour l’aide et le soutien
qu’elle m’a apportés durant ces trois années.
Que Madame Olivier-Bourbigou, chercheur à l’Institut Français du pétrole et
Messieurs Marc Lemaire, Professeur à l’Université Lyon 1, Bruno Chaudret, Directeur de
Recherche au CNRS, Jean-Pierre Simonato, chercheur au Commissariat à l’Energie
Atomique et David Cole-Hamilton, Professeur à l’Université de St Andrews, soient vivement
remerciés de l’honneur qu’ils m’ont fait en acceptant de juger ce mémoire.
Ce travail a été effectué en collaboration avec le Commissariat à l’Energie Atomique,
l’Institut Français du pétrole et la société Rhodia. Je tiens à remercier ces partenaires pour
l’aide qu’ils ont portée à la progression de cette recherche.
Je tiens également à remercier Monsieur Bernard Fenet, le professeur Denis Bouchu,
le professeur Agilio Padua pour leur aide .
Que tous les membres du laboratoire trouvent dans ces quelques lignes l’expression de
ma plus profonde gratitude. Je pense en particulier aux étudiants en thèse ou en DEA avec
lesquels j’ai partagé tout ou une partie de ces trois années.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
1
/
175
100%