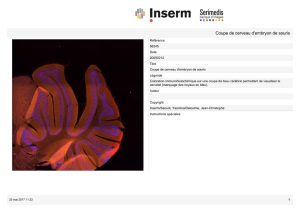
actualités - Société Française d`Immunologie

S
OMMAIRE
actualités
Société Française d’Immunologie
2012 N°126
E
ditorial
Par Roland Liblau, Président de la SFI
Juillet
E
ditorial
p.1
Conseil d’administration
Renouvellement p.2
Prix Jacques Oudin
Appel à candidature p.6
Brèves
1/La sérotonine est impliquée dans la
différenciation des ostéoclastes p.8
2/ Le TLR5 : un partenaire crucial pour la
phogocytose et l’éliminatioon de P. Aeruginosa
par les macrophages alvéolaires impliquant
l’IL-1b et l’asparagine endopeptidanse p.10
3/ Souris humanisées pour le système
immunitaire : un outil multidimmentionnel
et évolutif pour la recherche biomédicale
humaine p.13
4/ GADD34 régule la production de cytokines
en réponse au ARN double-brin : un lien entre
réponse au stress et immunité innée p.17
5/Activation de l’alarmine IL-33 par les
protéases des neutrophiles p.20
6/ Programmés pour tuer : les lymphocytes T
CD8+ mémoires potentialisent les activités
microbicides des monocytes inflammatoires
et des neutrophiles lors d’une réinfection p.22
7/ Cancer du sein métastatique : la lympho-
divpénie permet de pronostiquer la survie
globale p.25
Cours
23e Cours Annuel p.28
Clubs
4e workshop CFNI p.30
8e Colloque de Vaccinologie p.32
16e Colloque Cytokines et Chimiokines p.34
M
eetings
Congrès Européen d’Immunologie p.36
Bourses et Prix p.37
EFIS/Biolengend Young Investigator Award p.37
Congrès International d’Immunologie p.40
SFI
La vie de la SFI en 2012 et 2013 p.41
Chers amis,
sein de nos laboratoires. Certes, les appels
récompensé de grands projets portés par
des immunologistes aussi bien à Paris qu’en
ministre et son cabinet.
partenaires de l’Immunologie médicale. Il
La France y est bien représentée et des
a permis de resserrer les liens qui nous
programme remarquable et le Colloque
Cours Annuel de la SFI aura lieu comme
sur le site de la SFI.
depuis de nombreuses années.

SFI Actualités 2
notre Société lors de sa réunion de
du Conseil.
ligne. Nous nous sommes assurés de
2012 à midi.
conseil d’administration
in situ les réponses biologiques des
comprendre comment les lymphocytes T
mécanismes par lesquels ils agissent
Je souhaite aujourd’hui rejoindre le
pour notre discipline.
J’ai l’honneur de me porter candidat au
d’années.
Président :
Roland Liblau*
Toulouse
Vice-Présidente :
Anne Hosmalin
Secrétaire Général :
Eliane Piaggio
Trésorier :
Jean-Luc Teillaud
Cordeliers, Paris
Conseillers :
Olivier Boyer
Sylvia Cohen-Kaminsky
Sophie Ezine
Jean Pierre Farcet
Pierre Ferrier
Sylvain Fisson
Sylvie Fournel
Joel Pestel
Corinne Tanchot
Abdelilah Wakkach
Tours
*Les noms en orange indiquent conseilliers sortants

SFI Actualités 3
monoclonal. J’ai par ailleurs dirigé 6
plusieurs projets de recherche ainsi
bourses doctorales ou post doctorales.
jeunes talents.
de la Société Française d’Immunologie
années, je suis impliqué dans l’étude des
cancers.
département d’enseignement et aussi
du master d’immunologie.
président de la CSS7.
Société Francophone de Thérapie
Les raisons qui m’ont conduit à
discipline pour les plus jeunes qu’ils
ou médicale.
classe,
depuis 2007.
Actuellement, je suis intéressé par

SFI Actualités 4
Laboratoire d’Immunologie
Laboratoire d’Immunologie
Je souhaite aujourd’hui présenter ma
de la Société Française d’Immunologie
de notre discipline, comme je le
du trophoblaste dans la tolérance de
immunitaire maternel. J’ai obtenu mon
l’Inserm, je suis membre de la SFI
depuis de nombreuses années. J’ai
de recherche dans l’équipe que je
comprendre les mécanismes de
tolérance périphérique, en étudiant
la souris. Notre unité de recherche
d’enseignement dans des masters
d’immunologie.
annuel est une occasion unique de
Je suis impliqué dans plusieurs groupes
je suis actuellement secrétaire le
Sclérodermie, dont je suis trésorier
notre Société, sensible à la nécessité
biologistes et chercheurs, et suis
l’occasion pour les plus jeunes d’assister
connaître dans notre communauté. Les
cours annuels de la SFI et les réunions
organisées par les clubs sous la tutelle
de la SFI sont également des occasions
et rayonnement de l’immunologie en
beaucoup d’entre nous rencontrons en
ce moment.
la région Parisienne, il m’en reste
Française d’Immunologie SFI et je
présente ma candidature au Conseil
Cochin
Je suis membre de la Société Française
je souhaite maintenant prendre une part
clinique surtout orientée dans la
au départ sur l’étude du répertoire des
sur l’autoimmunité humorale et son
banques de cellules, plasmas, sérums

SFI Actualités 5
est d’étudier comment les récepteurs à
au sein du Laboratoire d’Immunologie
incluant notamment l’Immunologie
au CA de la SFI s’inscrit résolument
oncologie
en tant que membre de la Commission
L’immunothérapie a déjà un impact
la progression tumorale est encore
étapes moléculaires et cellulaires qui
conduisent à une réponse immunitaire
appropriés, d’élaborer des stratégies
Nous nous concentrons tout
cellules cancéreuses mais pas dans
immunogénicité remarquable, est au
cœur du programme de recherche
Notre programme de recherche est
croissante de l’immunothérapie et jouer
d’Immunologie, je souhaite contribuer
dans ma spécialité, l’immunologie des
tumeurs, en France.
FACEBOOK
également sur le réseau
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
1
/
40
100%