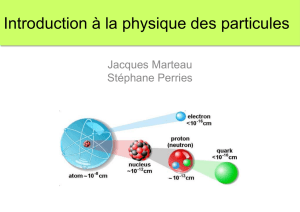
II. Propriétés diélectriques - Accueil thèses

T
TH
HÈ
ÈS
SE
E
En vue de l'obtention du
D
DO
OC
CT
TO
OR
RA
AT
T
D
DE
E
L
L’
’U
UN
NI
IV
VE
ER
RS
SI
IT
TÉ
É
D
DE
E
T
TO
OU
UL
LO
OU
US
SE
E
Délivré par l'Université Toulouse III - Paul Sabatier
Discipline ou spécialité : Sciences et Génie des Matériaux
JURY
Dr. F. Bauer (Rapporteur)
Dr. G. Seytre (Rapporteur)
Dr. J-M. Bergerat (Examinateur)
Pr. A. Soum (Examinateur)
Pr. C. Lacabanne (Directeur de thèse)
Dr. E. Dantras (Directeur de thèse)
Ecole doctorale : Science de la matière
Unité de recherche : CIRIMAT/Laboratoire de Physique des Polymères
Directeur(s) de Thèse : Pr. C. Lacabanne, Dr. E. Dantras
Rapporteurs : Dr. F. Bauer, Dr. G. Seytre
Présentée et soutenue par Jean-Fabien CAPSAL
Le 23 Octobre 2008
Titre : ELABORATION ET ANALYSE DES PROPRIETES PHYSIQUES DE NANOCOMPOSITES
HYBRIDES FERROELECTRIQUES

Ce travail de thèse a été réalisé au Laboratoire de Physique des Polymères/CIRIMAT,
Université Paul Sabatier/Toulouse. Il a bénéficié du support de la DGE et de la Région Midi-
Pyrénées, dans le cadre du programme NACOMAT.
Je tiens à remercier M. Alain Soum, Professeur à l’Ecole Nationale Supérieure de Chimie et
Physique de Bordeaux qui m’a fait l’honneur de présider ce jury de thèse.
M. Gérard Seytre, Directeur de recherche CNRS au Laboratoire des Matériaux Polymères et
des Biomatériaux de l’Université Claude Bernard de Lyon, a accepté d’examiner et de juger
ce travail de thèse. Je tiens à lui exprimer ma sincère reconnaissance.
M. François Bauer, Docteur d’état et Président de Piezotech S.A., m’a fait l’honneur d’être
rapporteur de ce travail de thèse, qu’il trouve ici l’expression de ma profonde reconnaissance.
M. Jean-Michel Bergerat, Docteur, responsable Recherche Amont « Matériaux et Procédés-
Composites » à Airbus, a coordonné les tâches « Détection de chocs » et « Dégivrage » du
programme NACOMAT. Il m’est agréable de le remercier pour son efficacité et sa
bienveillance tout au long de cette recherche.
J’exprime mes remerciements au Professeur Colette Lacabanne et au Docteur Eric Dantras
pour m’avoir accueilli dans leur groupe de recherche ainsi que pour leurs conseils avisés et
leur disponibilité. Qu’ils trouvent ici ma profonde et sincère reconnaissance.
Merci à Jany Dandurand, Sabastien Racagel, Mathieu Chevalier et Antoine Lonjon pour
l’aide qu’ils m’ont apportée et les nombreuses discussions que nous avons échangées.

Sommaire
Introduction 1
Chapitre 1 : Synthèse Bibliographique 4
I. Introduction 5
II. Matériaux ferroélectriques 6
II.1. Matériaux inorganiques 6
II.2. Matériaux organiques 7
II.3. Protocoles de polarisation 11
III. Propriétés physiques des composites hybrides 13
III.1. Connectivité 13
III.2. Transition vitreuse 14
III.3. Propriétés mécaniques 16
a) Résultats 16
b) Modélisation 18
Lois de mélanges 18
Méthodes d’homogénéisation 19
III.4. Propriétés diélectriques 20
a) Résultats 20
b) Modélisation 22
Modèle de milieu effectif 22
Modèle de Lichtnecker 24
III.5. Propriétés électroactives 24
a) Résultats 24
Effet piézoélectrique et pyroélectrique direct 24
Effet piézoélectrique inverse et électrostriction 28
b) Modélisation 29
Modèle de Furukawa : composites de connectivité 0-3 29
Modèle de connectivité mixte.......................................................................................32

Sommaire
Chapitre 2 : Matériaux et Méthodes 43
A. Matériaux 44
I. Structure des matériaux constitutifs 44
I.1. Polyamide 11 44
a) Structure chimique 44
b) Stabilité chimique 45
c) Structure physique 45
I.2. Copolymères P(VDF-TrFE) 46
a) Structure chimique 46
b) Stabilité chimique 46
c) Origine de la ferroélectricité des copolymères fluorés 46
I.3. Titanate de Baryum 47
a) Structure cristalline 47
b) Transition de phase cristalline 48
c) Synthèse et granulométrie 49
d) Structure cristalline des nanoparticules 50
II. Elaboration des nanocomposites 51
II.1. Protocole d’élaboration 51
a) Matrice polyamide 11 51
b) Matrice P(VDF-TrFE) 52
II.2. Etude de la dispersion des n-BT 52
II.3. Détermination de la fraction volumique 56
II.4. Protocole de polarisation 59
a) Matrice passive : polyamide 11 60
b) Matrice active : P(VDF-TrFE) 60
B. Méthodes 61
I. Analyse calorimétrique diatherme (ACD) 61
I.1. Matériaux organiques 62
a) Polyamide 11 62
b) P(VDF-TrFE) 64

Sommaire
I.2. Matériau inorganique : Titanate de Baryum 65
II. Analyse mécanique 66
II.1. Essais mécaniques en traction 66
a) Dispositif expérimental 67
b) Polyamide 11 67
II.2. Analyse mécanique dynamique (AMD) 68
a) Principe 69
b) Dispositif expérimental 69
c) Mise en œuvre des échantillons 71
d) Comportement dynamique mécanique du polyamide 11 71
III. Analyse diélectrique 72
III.1. Origine de la polarisation 72
a) Approche macroscopique 72
b) Approche microscopique 73
c) Origine des différentes polarisations 74
III.2. Phénomènes de relaxations 75
a) Equation de Debye 75
b) Equation d’Havriliak-Negami 76
III.3. Dépendance en température des temps de relaxation 77
a) Comportement de type Arrhenius 77
i) Théorie des barrières 78
ii) Phénomène de compensation 79
iii) Critère de coopérativité de Starkweather 80
b) Comportement de type Vogel-Tammann-Fulcher 81
i) Théorie du volume libre 82
ii) Théorie d’Adam-Gibbs 82
III.4. Spectroscopie diélectrique dynamique 83
a) Matériaux organiques : mobilité moléculaire 84
i) Polyamide 11 84
iii) P(VDF-TrFE) 88
iv) Conductivité et phénomènes inter-faciaux 90
b) Matériau inorganique 91
i) Relaxations solides-solides des nano-BT 91
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
1
/
267
100%