PSYCHIATRIE

1
HÉMATOLOGIE
LE SANG ET LE SYSTÈME LYMPHATIQUE
I - LA FORMATION DU SANG
A - LES CARACTÉRISTIQUES PHYSIQUES DU SANG
Phase cellulaire :
Globules rouges
Leucocytes
Plaquettes
Phase plasmatique :
Plasma : protéines de la coagulation
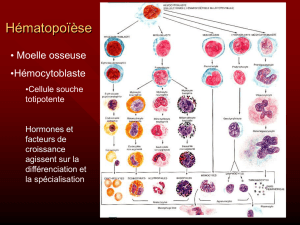
B - L’HÉMATOPOÏÈSE
Les cellules sanguines sont fabriquées dans la moelle osseuse rouge.
A partir d’une cellule souche commune pluripotente.
Cette cellule souche va se différencier vers :
Une cellule souche myéloïde
Une cellule souche lymphoïde
C’est l’hématopoïèse.
1) LA CELLULE SOUCHE MYÉLOÏDE
Elle se différencie à son tour en se multipliant :
a) Lignée érythroblastique : globules rouges
Les érythroblastes, ou hématies, ou globules rouges mûrissent progressivement en prenant différentes
formes.
Ils ne passent de la moelle osseuse dans le sang que lorsqu’ils sont matures.
Ce sont alors des cellules sans noyau : réticulocites.
Ils contiennent encore un peu d’ARN.
Ensuite ils deviennent des globules rouges : érythrocites.
La numération des réticulocites, ou globules rouges jeunes, indique la production médullaire.
La durée de vie d’un globule rouge est de 120 jours.
b) Lignée granuleuse : globules blancs
C’est elle qui va donner les leucocites.
Eux aussi ne passent dans le sang que lorsqu’ils sont matures :
Polynucléaires basophiles
Polynucléaires éosinophiles ou acidophiles
Polynucléaires neutrophiles
Monocytes
c) Lignée mégacariocytaire : plaquettes
Ce sont les mégacaryocytes.
Cellules qui se multiplient sur elles-mêmes : endomitose.
Elles multiplient leurs chromosomes.
Cela donne de grosses cellules avec beaucoup de chromosomes et un énorme cytoplasme.
Les plaquettes sont des fragments cytoplasmiques de ce mégacaryocyte une fois qu’il a mûri.

2
2) LA CELLULE SOUCHE LYMPHOÏDE
Elle provient de la même cellule souche pluripotente.
a) Lymphocytes B
Une partie des cellules lymphoïdes se développe au niveau de la moelle osseuse :
Ce sont les lymphocytes B.
b) Lymphocytes T
Une autre partie se développe et se différencie au niveau du thymus :
Ce sont les lymphocytes T.
3) FACTEURS NECESSAIRES A L’HEMATOPOÏESE
a) Les vitamines
Il est besoin de deux vitamines essentielles pour que l’hématopoïèse se fasse correctement :
Vitamine B12
Sa carence entraîne la maladie de Birmer
Folates : apportés par l’alimentation (légumes)
Leur carence entraîne une maladie au niveau de la moelle osseuse.
b) Les facteurs de croissance hématopoïétiques
IL3 : multi CSF (Colonie Stimulatif Facteur)
GM-CSF : granuleux monocytes
G-CSF : granuleux
M-CSF : macrophages
IL5 : polynucléaires éosinophiles
IL6 : mégacaryocytes
Érythropoïétine : protéine qui stimule la fabrication des globules rouges par la moelle.
Elle est fabriquée par les reins.
Injections sous-cutanées en cas d’insuffisance rénale (après chaque diurèse).
Cellules recombinantes identiques à la cellule humaine : fabriquées par le génie génétique.
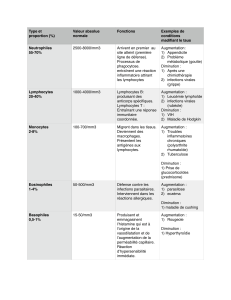
4) LA LIGNEE ERYTHOBLASTIQUE
Voir tableau
II - LES ELEMENTS FIGURES DU SANG
A - LES GLOBULES ROUGES
1) DESCRIPTION
Cellules biconcaves.
Ils possèdent une membrane très flexible qui leur permet de passer dans les petits capillaires.
Le durcissement de cette membrane provoque des ischémies.
Les globules rouges n’ont pas de noyau.
La sphérocytose est une maladie de la membrane qui la fragilise.
La rate les détruit alors plus facilement.
Leur rôle est de transporter l’oxygène vers les tissus.
Cela se fait grâce à la principale molécule qu’ils contiennent : l’hémoglobine (Hb).
L’anémie est une diminution du taux d’hémoglobine dans le sang.
Chez un sujet normal :
Hb A1 majoritaire (97%) 2 et 2.
Un peu d’Hb A2 (3%)
Traces d’Hb F (fœtale)

3
2) PATHOLOGIES
a) La thalassémie
C’est une anomalie génétique de la répartition des chaînes polypeptidiques et de l’hémoglobine.
b) La drépanocytose
C’est une anomalie de l’hémoglobine :
Mutation complète de l’Hb qui rigidifie les globules rouges.
Elle provoque des ischémies douloureuses.
3) LE ROLE DU FER
Il est absorbé par le duodénum.
Il provient de l’alimentation : viandes, légumes (moins bien absorbé).
4) LA MEMBRANE
a) Les antigènes
Ils se trouvent sur la membrane.
Il arrive qu’on se mette à fabriquer des anticorps contre ses propres antigènes :
Anémie hématopoïétose auto-immune.
b) Les enzymes
Ils métabolisent le glucose.
G6PD : pathologie fréquente de déficit de ce G6PD.
Pyruvate-kinase
B - LES GLOBULES BLANCS ou LEUCOCYTES
1) LES LEUCOCYTES GRANULEUX
a) Les polynucléaires neutrophiles
Défense contre les agressions bactériennes.
Ingestion par phagocytose.
Digestion/destruction : bactéricidie.
Durée de vie : 6 à 12 heures.
b) Les polynucléaires basophiles
Réaction d’hypersensibilité : allergies.
Libération d’histamine.
c) Les polynucléaires éosinophiles
Réaction d’hypersensibilité : défense parasite.
Phagocytose : bactéricidie.
d) Les monocytes
Phagocytose des éléments étrangers du sang.
Présentation des antigènes aux lymphocytes qui vont produire des anticorps pour détruire les bactéries.
Fabrication :
D’interféron
De monokine
De TNF
2) LE SYSTEME LYMPHOÏDE
a) Les lymphocytes T
80% des lymphocytes.
Deux grandes familles :
T4 helpers : ils aident à la réaction immunitaire
T8 : suppresseurs cytotoxiques (majoritaires)
Récepteurs spécifiques.
b) Les lymphocytes B

4
5 à 15% des lymphocytes circulant.
Fabrication des anticorps : plasmocytes.
Présents dans :
Le sang
La moelle
Les ganglions lymphatiques
Dans les ganglions, la réaction immunitaire stimule les lymphocytes.
Elle les transforme en plasmocytes : lymphocytes stimulés pour fabriquer des anticorps.
3) LES ANTICORPS
Ils sont produits par les lymphocytes B.
Ce sont des immunoglobulines (Ig).
Il en existe 5 classes.
Chacune ayant une chaîne lourde et une chaîne légère.
a) Chaîne lourde :
Ig M : chaîne lourde de type
Ig D : chaîne lourde de type
Ig G : chaîne lourde de type
Ig A : chaîne lourde de type
Ig E : chaîne lourde de type
D’abord apparaissent les anticorps Ig M.
Puis les Ig G montent et les Ig M descendent.
Les Ig G persistent tout au long de la vie : signe d’immunisation.
b) Chaînes légères (kapa) et (lambda).
Dans les conditions normales, on fabrique les deux familles.
E - ORGANISATION DE LA DEFENSE IMMUNITAIRE
1) CIRCULATION DES LYMPHOCYTES
a) Organes de prolifération
Moelle osseuse Sang Tissu ganglionnaire
Essentiellementles ganglions.
Un peu de tissu lymphoïde des organes :
Rate
Voies respiratoires
Tube digestif
Amygdales
Les lymphocytes repartent du tissu ganglionnaire par les vaisseaux lymphatiques.
Ils rejoignent le canal thoracique qui se jette dans le cœur.
b) Lymphocytes T
Régulateurs de la réponse immunitaire.
Antigènes associés aux molécules HLA qui se trouvent sur toutes les cellules du corps.
c) Lymphocytes B
Ig de surface.
Lymphocytes de mémoire : cellules B mémoire :
Reconnaissance de l’antigène
Prolifération
Fabrication d’anticorps
F - L’HEMOSTASE

5
Étude de la coagulation du sang.
Deux phases :
1) L’HEMOSTASE PRIMAIRE : PHASE CELLULAIRE
Quand les capillaires sont coupés, il y a lésion vasculaire.
Tout l’arbre circulatoire est recouvert d’une monocouche de cellules endothéliales internes qui empêche le
sang de coaguler à son contact.
Si on coupe cette couche endothéliale, le sang vient au contact des couches sous-endothéliales.
Les plaquettes adhèrent (se collent) au niveau du sous-endothélium.
Elles contiennent un facteur fabriqué par les cellules endothéliales et nécessaire à l’adhérence des
plaquettes : le facteur de Willebrand.
Les plaquettes s’agrègent : agrégation plaquettaire.
On mesure le TS : temps de saignement.
Son augmentation est symptomatique d’un trouble de l’hémostase primaire :
Thrombopénie : manque de plaquettes
Thrombopathie : maladie des plaquettes
Maladie de Willebrand : non adhérence des plaquettes
2) PHASE PROTÉIQUE : COAGULATION PROPREMENT DITE
Les facteurs de coagulation, qui sont des protéines, sont fabriquées par le foie.
La cirrhose ou l’hépatite aiguë entraînent une diminution de la coagulation.
Système régulateur.
Système enzymatique.
Les protéines vont s’activer les unes les autres.
Deux voies pour activer la coagulation :
a) Système endogène
Contact avec une surface mouillable : structures sous-endothéliales.
1er facteur activé : facteur XII
2ème : facteur XI
3ème : facteur IX (facteur antihémophilique B)
Le facteur IX s’associe au facteur VIII pour donner un complexe enzymatique (FAHA).
Besoin de calcium et de phospholipides qui proviennent de la membrane plaquettaire des plaquettes
activées.
Les deux phases sont interdépendantes.
L’ensemble active le facteur X.
Le facteur X s’associe au facteur V, plus des phospholipides et du calcium pour donner un nouveau
complexe enzymatique.
Celui-ci active le facteur II.
Facteur II activé = Thrombine : grosse molécule
Facteur II inactivé = Prothrombine : molécule tronquée
Ces molécules circulent dans le sang.
La thrombine transforme le fibrinogène en fibrine (facteur I) qui forme le caillot.
L’Actilyse casse la fibrine.
b) Système exogène
Le facteur VII est activé par le facteur tissulaire qui provient des tissus lésés, abîmés par un traumatisme.
Le facteur VII activé va activer à son tour le facteur X qui rentre dans le complexe enzymatique déjà formé
par le système endogène.
c) La thrombine est une enzyme puissante
Principal facteur de la coagulation.
La thrombine active à son tour le facteur V et le facteur VIII.
 6
6
1
/
6
100%