P2-UE9-Gasque-Maladies_auto-immunes-_Partie

UE9 – Immunopathologie et Immunointervention
Pr P Gasque
Date : 18/03/16 Plage horaire : 8h30-10h30
Promo : DFGSM2 2015/2016 Enseignant : Pr Gasque
Ronéistes :
NATIVEL Mathilde
CHANE-CHAN Amélie
Mécanismes physiopathologiques des maladies auto-
immunes (MAI) - Partie 1
I. Introduction: Définition et concepts clés
II. Deux types de MAI (SO et NSO)
III. Mécanismes de tolérance (Centrale et périphérique des
lymphocytes T et B)
IV. Rupture de la tolérance et autoréactivité
A. Modification de l’autoantigène (médicament)
B. Réaction croisée entre pathogène et Ag du soi
C. Libération des Ags séquestrés (et modifiées)
V. Dysfonctionnement des T regs
VI. Autres facteurs favorisant l’auto-immunité
VII. Mécanismes d’action des autoanticorps
A. Modulation de l’activité des récepteurs
B. Cytotoxicité
VIII. Mécanismes d’action des cellules T cytotoxiques
IX. Traitements des MAI
Les diapos sont à la fois en anglais et en français afin de nous familiariser avec les terminologies anglaises.
Objectif : obtenir les bases de l’auto-immunité et les illustrer avec certaines pathologies tels que le lupus, la
polyarthrite rhumatoïde ou encore la sclérose en plaque.

I. Introduction: Définition et concepts clés
Dans l’immunologie, il existe une « face » qui est protectrice et une autre plutôt pathologique, dont l’auto-
immunité fait partie. Dans ce cours, nous verrons les éléments clés de cette auto-immunité et des maladies
auto-immunes (MAI), avant d’aborder plus tard des éléments plus spécifiques des MAI telles que la sclérose
en plaque, de la polyarthrite rhumatoïde ou encore le lupus.
Tout d’abord, l’auto-immunité signifie au moins l’immunité classique, sauf qu’au lieu d’être dirigée vers un
virus, un parasite ou une bactérie, elle est dirigée contre une cellule du Soi. Il y aura donc un antigène du
Soi, qu’on appellera un auto-antigène, qui sera reconnu soit par des autoanticorps soit par des LT
autoréactifs.
Pendant ce cours, on verra que, fort heureusement, ce n’est pas systématique. La grand majorité d’entre
nous, avons des systèmes régulateurs de notre système immunitaire qui vont nous empêcher de réagir contre
nos propres composants. Cependant, chez certains patients, (compte tenu de la génétique, du système HLA,
des facteurs environnementaux etc.) cette balance et ce contrôle du système immunitaire ne s’exerce pas
suffisamment et on aura alors un phénomène de ??? , une perte de rétrocontrôle indiquée ci-dessus en
jaune.
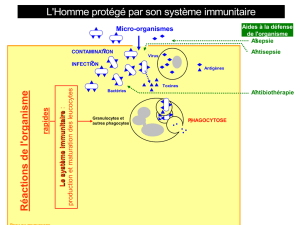
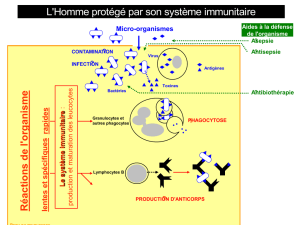
On va rappeler le rôle des lymphocytes T, B qui
sont dirigés contre nos propres cellules et le rôle
clé de la cellule présentant l’antigène.
Il faut que vous les reconnaissiez ici par scan
électro-microscopique. Les LT et LB vont recevoir
une instruction de la cellule présentant l’antigène
(en anglais « Antigen presenting cell ») et ces 3
coupables agissent contre nos propres cellules, les
cellules du Soi.
Définition : L’auto-immunité concerne toute réaction immunitaire cellulaire (lymphocytes T ou B
autoréactifs) ou humorale (production d’auto-anticorps) développée vis-à-vis des propres constituants de
l’organisme.
Les réponses auto-immunes sont dirigées contre
des éléments (antigène) du soi appelé auto-
antigène (Exemples: acides nucléiques :
AN/lupus ou encore récepteur de l’acétylcholine,
neurotransmetteur: Myastenia gravis).
L’auto-immunité peut être spécifique d’un organe (MAI SO) (thyroïde: hashimoto) ou systémique (MAI
NSO = non spécifique d’organe, lupus).
Il existe normalement des mécanismes de tolérance immunitaire mais qui font défauts dans les MAI.
Les MAI sont des pathologies chroniques (Exple: sclérose en plaques).
On peut contrôler (par médicament, en contrôlant la concentration d’antigène, en essayant même de les
éliminer) mais on ne peut jamais guérir d’une MAI.

Voyons quelques exemples de maladies auto immunes,
dont 2 qui illustrent particulièrement bien le propos : le
vitiligo et la sclérose en plaque
On peut lire sur la diapositive ci-contre la traduction de
ces différentes maladies en Anglais. Il est en effet
souhaitable que nous futurs médecins soyons capables de
communiquer dans cette langue résolument swagg.
Le vitiligo (visible à l’oeil nu): maladie qui va atteindre la peau et qui va toucher particulièrement les
mélanocytes. Le phénomène de dépigmentation visible sur cette jeune patiente est donc dû dans cette
pathologie auto-immune à la destruction des mélanocytes (qui produisent de la mélanine, ensuite absorbée
par les kératinocytes, qui et qui justifie la coloration brune de la peau). Ce n’est pas la MAI la plus
douloureuse au sens analgésique du terme, mais elle a un fort impact sur la vie sociale du patient.
Ce qui est représenté en (b) et en (c), c’est le nombre de cellules qui
au départ qui vont « infiltrer » (en marrons) et au-dessus il y a la
peau. Les mélanocytes sont indiqués par les flèches noires et très
rapidement ceux-ci vont disparaitre chez ce patient.
Mais il faut savoir que le mélanocyte se retrouve quasiment dans
tous les tissus, il fait partie des cellules nomades. Donc dans un
vitiligo, il n’y aura pas uniquement la peau qui présentera un
phénomène de destruction mais quasiment tous les tissus. La cible
du système immunitaire est une enzyme qui contribue à la synthèse
de mélanine à partir de la L-dopa appelée la tyrosinase. Il va y
avoir des auto-anticorps et des LT autoréactifs contre cette
tyrosinase, qui vont la détruire et par conséquent détruire les
mélanocytes.
La sclérose en plaque (pas visible mais détecté à l’IRM) va atteindre le SNC. On a une destruction de la
gaine de myéline des axones du SNC produite par les oligodendrocytes et pas celles du SNP. Les cellules
qui myélinisent le SNP sont les cellules de Schwann, qui concerne une autre pathologie auto-immune
appelée le syndrome de Guillain Barré.
Ici ce qui est indiqué en vert, ce sont ces très jolies cellules qui
sont capables de myéliniser les axones (en noir). Cette myéline
étant une gaine protectrice des axones qui aide à la conduction
de l’influx nerveux. C’est tout simplement comme si vous aviez
un fil électrique avec une gaine blanche autour.
Là, la myéline va empêcher l’influx nerveux de se répandre
partout et lui permettre d’être conduit d’un bout à l’autre de
l’axone sans coupure de signal. Elle est donc synthétisée par les
oligodendrocytes, notamment un de ces constituants : le MDP
pour Myelin Basic Protein. Ce dernier est malheureusement une
des cibles du système immunitaire chez ces patients.
La sclérose en plaque (Multiple Sclerosis) est donc due à une reconnaissance de la MBP ou de MOG
« Myelin Oligodendrocyte Glycoprotein ». Deux cibles (non uniques), qui font que, s’il y a des LT
autoréactifs ou des auto-anticorps, ils vont détruire la cellule qui exprime ces molécules à savoir les
oligodendrocytes.

Dans la myasthénie (Myastenia Gravis), il existe des anticorps dirigés contre un récepteur de
l’acétylcholine au niveau de la jonction neuro-musculaire qui va empêcher la transmission du signal
nerveux vers la plaque musculaire et engendrer une perte de la motricité
Le Diabète de Type I, important à distinguer du diabète de type II (qui n’est pas une MAI,) s’explique par
une production d’anticorps contre l’hormone insuline qui est produite par les cellules bêta du pancréas. Il
y’a donc destruction de ces cellules : perte de synthèse d’insuline : diabète, perte d’absorption du glucose.
Le Lupus va affecter essentiellement la peau, donc ça se voit assez facilement chez les patients atteints.
Tous les organes sont touchés car la cible du lupus c’est l’ADN et les protéines nucléaires exprimées dans
toutes les cellules avec des auto-anticorps et des lymphocytes T autoréactifs dirigés contre notre propre
ADN, ARN ou encore contre les ribonucléoprotéines.
La polyarthrite rhumatoïde (PR) (Rhumatoid arthritis RA) a pour cible les fibroblastes synoviaux. Les
auto-anticorps sont dirigés contre des protéines produites dans les fibroblastes, sauf que ces protéines sont
anormales car elles ont été modifiées par un processus appelé la citrullination où les arginines sont
remplacées par de la citrulline. Il y a donc des protéines citrullinées, qui n’existent normalement pas dans
la nature, reconnues par le système immunitaire comme éléments antigéniques, étrangers. S’en suit une
destruction de l’articulation puisque le tissus synovial y joue un rôle important pour la lubrification.
Tous les organes peuvent être le siège de
mécanismes auto-immuns comme on peut le
voir sur la diapo ci-contre, l’objectif n’étant
pas de faire de manière exhaustive le
catalogue de toutes ces pathologies mais de
comprendre les mécanismes essentiels.
Concept n°1 : L’immunité T et B antivirale est identique à la réponse auto-immune dirigé contre le soi.
Ce rappel sert à montrer 2 choses :
Les LT et les LB contribuent à la MAI de la
même manière qu’ils vont contrôler l’immunité
induit par un pathogène.
Les voies d’activation qui sont engagées, seront
des cibles potentielles dans les traitements.
Systématiquement, les traitements des MAI
aujourd’hui visent à inhiber les voies de
stimulation du système immunitaire, avec
néanmoins le risque des maladies infectieuses.
Si un patient souffrant d’une MAI reçoit trop de
substances qui inhibent son système
immunitaire, on va le mettre à risque de
développer une pathologie infectieuse.

Quand il y a un antigène, celui-ci est phagocyté par une cellule présentant l’antigène, puis présenté aux LTs
(qu’on appelle LT naïfs ou Th0) au travers de la molécule HLA de base 2 (c’est le signal 1). De plus, il existe
un faux signal au travers la molécule CD80/86 qui est reconnue par la molécule CD28 portée par ces LTs.
Les 2 signaux sont obligatoires pour instruire l’activation de ces LTs, pour qu’ils se différencient soit Th1
qui produisent beaucoup d’interferon gamma, soit en Th2 qui produisent beaucoup d’interleukine 4
(Cependant ils ne produisent pas uniquement soit l’un soit l’autre, mais les quantités sont différentes). En
fonction de l’antigène, les proportions de Th1 et de Th2 vont varier, et il aura même Th17 mais il ne rentre
pas dans les détails. (Note du ronéolecteur : la liste des LT auxiliaire est un contenu bonus)
Ces cellules vont ensuite, par voies de synthèse de cytokines (telles que IL-4 ou IL-6) induire l’activation des
LBs, qui eux vont pouvoir être aussi activé directement par l’antigène par le BCR (B-cell Receptor). Les LBs
vont donc avoir également 2 types de signaux (pour éviter que le système immunitaire s’emballe
systématiquement). 1 signal n’est pas suffisant pour activer une cellule, il en faut 2 (mécanisme de contrôle
et de préservation). Cela va alors induire la production d’anticorps, de CD8 etc.
Il y a également beaucoup de cytokines qui sont produites (il cite les cytokines du schéma): elles vont avoir
un rôle d’activation des cellules entre elles (d’où « interleukines » pour « inter-cellules »).
Important : c’est le même schéma lorsqu’il s’agit d’un auto-antigène. Donc pour contrôler l’auto-
immunité, il faudra contrôler Th0, Th2, Th1 etc
Concept n°2 : Les MAI sont chroniques, complexes, et elles évoluent
Les MAI auront des processus différents pour une même pathologie telle que la sclérose en plaque. Par
exemple, ici, certains patients auront une pathologie progressive (en bas à gauche), tandis que d’autres
feront une guérison-rechute ou « Relapsing-remitting » (en haut à gauche).
 6
7
8
9
10
11
12
13
14
15
16
17
6
7
8
9
10
11
12
13
14
15
16
17
1
/
17
100%