dip_syndromiques_lfb_DIP SYNDROMIQUES 290313

Pr. Jean-Louis Stéphan, Service de Pédiatrie - CHU Saint-Etienne
Cahier n°1


- 1-
Sommaire
Cahier n°1
01- Introduction à l’étude des DIP 3
Quand suspecter une anomalie
constitutionnelle de l’immunité ? 4
Comment affirmer simplement
l’existence d’un DIP ? 7
02- Les déficits syndromiques 14
L’anomalie de Di George 14
Le cartilage-hair hypoplasia 22
Le syndrome de Netherton 26
Les candidoses muco-cutanées
chroniques (CMC) 30
Les syndromes Hyper IgE (HIE) 31
Les syndromes d’instabilité
chromosomique ou réparatoses 44
03- Annexes 60
Glossaire 60
Valeurs de référence 76
04- Références 77

ADG : Anomalie de Di George (ou DG)
AFP : Alpha-fœtoprotéine
ALPS : Auto-immune Lymphoproliferative
Syndrome (syndrome lymphoprolifératif
avec auto-immunité)
APECED : Autoimmune Polyendocrinopathy -
Candidiasis - Ectodermal - Dystrophy
ARNr : ARN ribosomaux
ATM : Serine/threonine protein kinase
Ataxiatelangiectasia Mutated
BCG : Bacille de Calmette et Guérin
BrdU : Bromodéoxyuridine
CEREDIH : Centre de Référence Déficits
Immunitaires Héréditaires
CHH : Cartilage-hair Hypoplasia
CMC : Candidoses Muco-cutanées Chroniques
CMF : Cytométrie de Flux
CV : Charge Virale
DICV : Déficit Immunitaire Commun Variable
DIP : Déficits Immunitaires Primitifs
EBV : Epstein-Barr Virus
FISH : Hybridation in situ en Fluorescence
FRH : Fièvres Récurrentes Héréditaires
Greffe de MO : Greffe de Moelle Osseuse
HIE : Syndromes Hyper IgE
HSV : Herpes simplex virus
ICF : Déficit Immunitaire, Instabilité Centromérique,
anomalies Faciales
IPEX : Immune dysregulation, Polyendocrinopathy,
Enteropathy, X-linked
IRM : Imagerie par Résonance Magnétique
LEKTI : Lymphoepithelial Kazal Type related
Inhibitor
LT : Lymphocyte T
LTa : Lymphotoxine a
NK : Natural Killer
MBL : Mannose-Binding Lectin
MIM : Online Mendelian Inheritance in Man
MLPA : Multiplex Ligation-dependent Probe
Amplification
MNI : Mononucléose Infectieuse
MO : Microscopie Optique
Nbs : Syndrome de Nijmegen (Nijmegen Breakage
Syndrome)
nbt : Test au Nitrobleu de Tétrazolium
PCR : Polymerase Chain Amplification
PN : Polynucléaires
RA : Acide Rétinoïque
RMRP : Ribonuclease Mitochondrial RNA Processing
ROR : Retinoïd Related Orphan Receptor
SLP : Syndrome Lymphoprolifératif
SNC : Système Nerveux Central
snoRNAs : Small Nucleolar RNA
TDM : Tomodensitométrie
TRECS : T cell Receptor Excision Circles
UV : Ultra-violets
Ca : Calcémie
CRP : C-Réactive Protéine
Fe : Fer sérique
GB : Globules Blancs
G/l : Giga/litre
Hb : Hémoglobine
LCR : Liquide Céphalorachidien
Ly : Lymphocytes
Na : Natrémie
PLT : Plaquettes
PN : Polynucléaires Neutrophiles
VGM : Volume Globulaire Moyen
Termes de biologie
Termes de biologie
Liste des abréviations
Liste des abréviations
- 2-

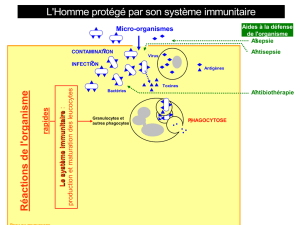
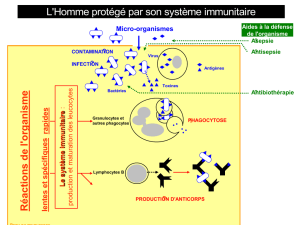
DEFAUTS GENETIQUES DE LA REPONSE IMMUNITAIRE
Introduction à l’étude des DIP
01
année avec un DIP et
qu’environ 5000 patients,
enfants et adultes
vivraient avec un tel
déficit.3,4
En outre, la prise en
charge a d’autant plus
de chance d’être efficace
qu’elle sera mise en place
précocement. C’est le cas
de la substitution par
immunoglobulines dans les anomalies de la fonction
humorale, de la greffe de moelle dans les déficits T
ou de l’antibiothérapie prophylactique. La correction
du défaut génétique par transfert de gène est même
désormais possible et en cours d’évaluation pour
un petit nombre d’entre eux.5
Avant d’illustrer les formes syndromiques de DIP*
qui constitueront l’objet de ce 1er cahier, quelques
« pistes » pour le diagnostic des DIP et des autres
erreurs innées de l’immunité seront brièvement
exposées.
Les anomalies constitutionnelles de
l’immunité sont multiples et varient dans
leur traduction clinique. Les déficits
immunitaires primitifs (DIP) sont
caractérisés par une atteinte qualitative
et/ou quantitative du système immunitaire,
entraînant une susceptibilité accrue aux
infections. Plus de 300 maladies sont
décrites et des mutations répertoriées
dans quelques 180 gènes, et de très
nombreux mécanismes impliqués
(cf répartition des différents DIP).1,2 Avec
les techniques de séquençage génome
entier et exome entier le rythme des
publications s'est accéléré et l'on peut
prédire l'élucidation de centaines de DIP
dans les 10 ans à venir (JL Casanova).1,2
Leur meilleure définition à l'échelon
moléculaire permet aujourd’hui d’envisager,
pour un grand nombre, un conseil génétique
et un diagnostic prénatal. La prévalence
dans la population générale est assez
mal connue et en France on estime
qu’environ 150 enfants naissent chaque
Le diagnostic des DIP
est une véritable urgence
car ces affections mettent
en jeu le pronostic
vital en raison
de la susceptibilité
aux infections ou
des manifestations
auto-immunes graves,
auxquelles elles exposent.
- 3-
Introduction
à l’étude
des DIP
55%22%
5%
10%
8%
Déficits humoraux
Déficits en lymphocytes T
Déficits en cellules phagocytaires
Anomalies du complément
Autres
Répartition des différents
DIP (2011) :
Données de la Société
Européenne des Déficits
Immunitaires -ESID-
http://www.esid.org/statistics.
php?sub=
* Cf. glossaire page 60
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
1
/
84
100%