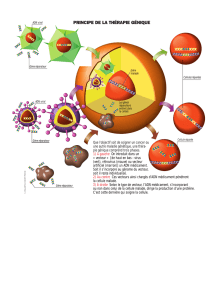
developpement d`un vecteur lentiviral spécifique

ALMA POSVANDZIC
DEVELOPPEMENT D'UN VECTEUR LENTIVIRAL
SPÉCIFIQUE AUX MACROPHAGES CÉRÉBRAUX
ACTIVÉS POUR LA THÉRAPIE GÉNIQUE
APPLIQUÉE À DES MALADIES DU SYSTÈME
NERVEUX
Mémoire présenté
à Ja Faculté des études supérieures de l'Université Laval
dans le cadre du programme de maîtrise en physiologie-endocrinologie
pour l'obtention du grade de Maître es Sciences (M.Se.)
FACULTE DE MEDECINE
UNIVERSITÉ LAVAL
QUÉBEC
DECEMBRE 2007
© Aima Posvandzic, 2007

Résumé
Le but général de ce projet de recherche était de valider la faisabilité de la méthode qui
consiste en l'introduction d'un gène extrinsèque dans les macrophages cérébraux à l'aide
d'un lentivirus, placé sous contrôle d'un promoteur connu pour être actif préférentiellement
dans ces cellules. Plus spécifiquement, nous avons : 1) identifié le gène de la galectine-3
comme étant exprimé de façon préférentielle dans les macrophages cérébraux activés dans
différentes conditions pathologiques du cerveau ; 2) caractérisé son promoteur ; 3) utilisé ce
promoteur pour la conception d'un vecteur lentiviral ; et 4) vérifié la possibilité d'utiliser
des cellules microgliales transduites comme vecteurs pour la thérapie génique. Nous avons
obtenu des résultats prometteurs quant à une utilisation potentielle des cellules microgliales
en thérapie génique ex vivo. Toutefois, pour confirmer que le vecteur lentiviral assure une
expression spécifique et durable du transgène dans un contexte inflammatoire, il faudrait
entreprendre d'autres analyses.

Avant-Propos
J'aimerais tout d'abord remercier mon directeur de recherche, le Dr Luc Vallières, pour ses
encouragements, ses précieux conseils, sa constante implication, sa grande disponibilité,
son enthousiasme contagieux, sa bonne humeur et bien plus encore. Vous avez ma plus
grande admiration et toute ma reconnaissance.
Je tiens aussi à remercier Pierrot Tremblay, notre précieux assistant de recherche, qui m'a
tout appris, ou presque, et m'a accordé tout au long de ma maîtrise une aide inestimable.
Un gros merci pour ta compétence, ta disponibilité et ton inépuisable sens de l'humour.
Je remercie chaleureusement tous mes coéquipiers, Hugo, Jérôme, Julie A., Caroline, Julie
P.,
Andréanne, Marie-Josée et Naika avec qui travailler était une pure joie, ainsi que mes
nombreux collègues de laboratoire que j'ai eu le bonheur de côtoyer quotidiennement.
Merci pour votre aide, vos sourires, vos fous rires et, surtout, votre amitié.
Merci à tous mes amis, d'ici et de loin, pour votre patience, support, encouragements,
amour et amitié qui m'ont aidé à aller au bout de ce projet. Je vous en serai toujours très
reconnaissante.
Finalement, et avant tout, je tiens à remercier mes parents et mon frère, pour leur soutien et
leur amour inconditionnel. Marna, tata i Alêne, volim vas najvise na svijetu !

À ma grand-mère Paula

Table des matières
Résumé i
Avant-Propos ii
Table des matières iv
Liste des abréviations vi
Liste des figures ix
Liste des tableaux x
Chapitre
1
: Introduction 1
1.1 Thérapie génique 2
1.1.1 Historique 2
1.1.2 Thérapie génique et système nerveux central : problèmes et solutions 3
1.1.3 Transfert de gènes in vivo et ex vivo 4
1.1.4 Vecteurs de transfert en thérapie génique 5
1.1.4.1
Vecteurs non-viraux 6
1.1.4.2
Vecteurs viraux 7
1.1.4.2.1
Virus de l'herpès simplex de type
1
(HSV-1) 7
1.1.4.2.2
Adénovirus 8
1.1.4.2.3
Virus adéno-associé (AAV) 9
1.1.4.2.4
Rétrovirus 10
1.1.4.2.5
Lentivirus 12
1.1.4.2.5.1
Génome lentiviral 12
1.1.4.2.5.2
Avantages et limitations des vecteurs lentiviraux 14
1.1.4.2.5.3
Production de vecteurs lentiviraux 15
1.1.4.2.5.4
Pseudotypage des lentivirus 17
1.2 Macrophages cérébraux 17
1.2.1 Origine et caractéristiques des macrophages cérébraux 18
1.2.2 Rôle des macrophages cérébraux dans des maladies du système nerveux 19
1.2.2.1
Macrophages et tumeurs cérébrales 21
1.2.3 Thérapie cellulaire des maladies du système nerveux 22
1.2.3.1
Macrophages cérébraux comme véhicules cellulaires dans la thérapie génique
des maladies du système nerveux 23
1.3 Utilisation d'un promoteur spécifique aux macrophages cérébraux pour la thérapie
génique 24
1.3.1 Galectine-3 24
1.3.2 Fonctions biologiques de la galectine-3 25
1.3.2.1
Galectine-3 et inflammation 26
1.3.3 Galectine-3 dans le cerveau 27
1.4 Objectifs du projet 28
Chapitre 2 : Matériel et méthodes 29
2.1 Étude de l'expression de la galectine-3 29
2.1.1 Hybridation in situ 29
2.1.2 Immunofluorescence 29
2.1.3 RT-PCR 30
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
1
/
75
100%