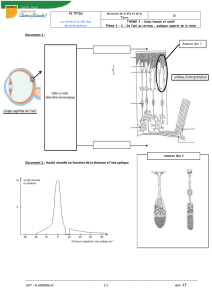
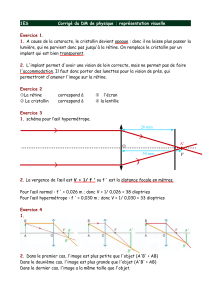
Vitréorétinopathie de transmission autosomique dominante : étude

1
UNIVERSITE PARIS VAL-DE-MARNE
FACULTE DE MEDECINE DE CRETEIL
!!!!!!!!!!!!!!!!!!!!
ANNEE 2007-2008 N°
THESE
POUR LE DIPLOME D’ETAT
DE DOCTEUR EN MEDECINE
Discipline : Ophtalmologie
------------------
Présentée et soutenue publiquement le 3 octobre 2008
A la faculté de médecine de Paris V René Descartes
------------------
Par Amandine BARJOL
Née le 31 Octobre 1979 à Vitry-Sur-Seine
------------------
Vitréorétinopathie de transmission autosomique dominante :
étude du phénotype dans une grande famille de patients
présentant une mutation du gène associé à la maladie de
Wagner.
PRESIDENT DE THESE : LE CONSERVATEUR DE LA
Monsieur le Professeur Antoine Brézin BIBLIOTHEQUE UNIVERSITAIRE
DIRECTEUR DE THESE :
Madame le docteur Sophie Valleix
Signature du Président de thèse Cachet de la
bibliothèque universitaire

UNIVERSITE PARIS VAL-DE-MARNE
FACULTE DE MEDECINE DE CRETEIL
!!!!!!!!!!!!!!!!!!!!
ANNEE 2008 N°
THESE
POUR LE DIPLOME D’ETAT
DE DOCTEUR EN MEDECINE
Discipline : Ophtalmologie
------------------
Présentée et soutenue publiquement le 3 octobre 2008
A la faculté de médecine de Paris V René Descartes
------------------
Par Amandine BARJOL
Née le 31 Octobre 1979 à Vitry-Sur-Seine
------------------
Vitréorétinopathie de transmission autosomique dominante :
étude du phénotype dans une grande famille de patients
présentant une mutation du gène associé à la maladie de
Wagner.
.
PRESIDENT DE THESE : LE CONSERVATEUR DE LA
Monsieur le Professeur Antoine Brézin BIBLIOTHEQUE UNIVERSITAIRE
DIRECTEUR DE THESE :
Madame le docteur Sophie Valleix
Signature du Président de thèse Cachet de la
bibliothèque universitaire

4
A Monsieur le Professeur Antoine Brézin,
Vous me faites l’honneur de présider ce jury. Pour
l’attention et la bienveillance dont vous m’avez encadrée
durant le stage effectué dans votre service chaleureux et
dynamique, soyez assuré de ma profonde admiration et
de ma sincère reconnaissance.

5
A Madame le Docteur Sophie Valleix,
Tu m’as fait l’honneur et la gentillesse de diriger ce
travail. Ta confiance et ta pédagogie m’ont guidée sur les
voies de la biologie moléculaire. Pour ta générosité et ta
passion, trouve ici le témoignage de toute mon amitié, et
de ma plus sincère reconnaissance.

6
A Madame le Professeur Pascale Massin,
Vous me faites l’honneur de juger ce travail. Pour la
richesse de l’enseignement que vous m’avez dispensé :
soyez assurée de ma sincère gratitude.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
1
/
118
100%