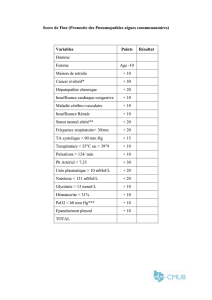
Un cas de suspicion de NFU1

R . M O N T O R I O L , S . R O C H E , E . L E B I G O T , J - C . N E T T E R ,
C . V I A N E Y - S A B A N , T . L E V A D E , A . B O U T R O N , P . B R O U E , F .
S A B O U R D Y
UN TROUBLE DE LA
LIPOYLATION
1

HISTOIRE DE LA MALADIE
•Née de parents non consanguins
•Déroulement de la grossesse et accouchement
normaux
•Suivie à Tarbes
•Stagnation staturo-pondérale
•Enfant grognon, irritable
•Lactates 4,1 mmol/L (N: 0,5 – 2)
2

HISTOIRE DE LA MALADIE
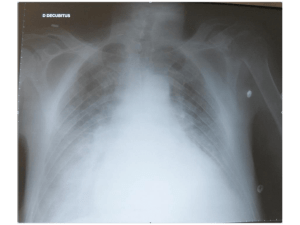
•Décompensation cardiaque droite
•Nécessitant prise en charge au CHU Toulouse
•Pneumocystose pulmonaire sur HTAP
•Echographie cardiaque
•LBA (Pneumocystis jiroveci)
3

EXAMEN CLINIQUE
•Retard staturo-pondéral
•Retard psycho-moteur
•Hypotonie axiale et périphérique majeure
•Pas de syndrome dysmorphique
4

BILAN DE 1ERE INTENTION
•Imagerie
•IRM cérébrale normale
•Échographie abdominale normale
•Scanner cardiaque: pas de malformation
•TGO 32 UI/L (N: 3 – 85)
•TGP 30 UI/L (N: 3 – 60)
•Bilan immunitaire normal
5
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
1
/
21
100%