Maladie de Huntington MH

Maladie de Huntington MH
Mise en evidence par un neurologue americain Hungtington en 1872 (avant le 19e siecle on les
persecutait car on pensait quils etaient possédés). Ds le depart, origine genetique suspectée. Il a
examine lhistoire medicale combinée de plusieurs generation dune meme famille avec les
symptomes.
Cest MND progressive et hereditaire conduisant a la mort entre 15 et 20 ans apres le diagnostic.
La duree devolution et le type de symptômes sont tres variables d'un patient a lautre et au sein
dune meme famille.
=> MND genetique. On parle aussi de choree (ensemble de symptome) de Hungtington ou de
danse de saint Guy.
I. Introduction
3 catégories de symptômes:
Cognitif (memoire, attention, planification)
Moteur (choree, dystonie, marche et equilibre)
Psychiatrique (depression, probleme de personnalite, psychose, conduite auto ou hetéro
aggressive).
On considere en general que les troubles cognitifs et moteurs saggravent au cours du temps mais
quils peuvent aussi se modifier alors que les troubles psychiatriques fluctuent au cours de la
maladie et peuvent apparaitre a nimporte quel moment de l'évolution. On trouve aussi un certain
nombre de troubles marginaux comme lepilepsie, les troubles du sommeil ou lincontinence.
=> Tres handicapante sur tous les plans de la vie quotidienne, professionnelle et sociale.
1. Troubles cognitifs
Trouble de la memoire, trouble de l'apprentissage et trouble du rappel améliore par indicage,
trouble de l'attention, trouble des fonctions exécutives, trouble du langage, trouble visuo spatiaux
(stades plus severes). Trouble cognitif discret voire inexistant chez certains patients au debut. Pour
certains auteurs peuvent également précéder les troubles moteurs. La progression de la MH ne
perturbe pas les fonction intellectuelles de maniere uniforme.
=> Lassociation trouble cognitif et trouble psychopathologique complique linterpretation et
levaluation des symptomes.
2. Troubles moteurs
Evolution lente avec symptomes subtils au depart. Certaines formes peuvent etre juveniles (avant
20ans) comme lepilepsie 3 a 16%, les myoclonies (mouvement anormaux) et la rigidite.
Choree: etat de mouvements spontanées, excessifs, irrégulièrement cadencés, distribues au
hasard et abruptes.
La severite peut varier dune legere agitation a un flot continu de mouvements invalidant tres

violent en passant par des exagerations intermittentes des gestes ou des expressions.
3. Troubles psychiatriques et comportementaux
Des le debut de la maladie dans1/3 des cas, desinsertion professionnelle et familiale qui a un
retentissement sur les performances cognitives. Leur frequence varie selon les etudes entre 35 et
73% au cours de levolution de la maladie (depression 9 a 41%, manie 10%, dysthimie 5 a 9%,
hallu 1, 6%, taux de suicide eleve 1 a 5, 7%, trouble des conduites sexuelles 25% et conduites
addictives).
Ne se declare pas immédiatement chez le sujet atteint qui a une vie normale avec sa declaration.
Symptomes typiques habituellement entre 30 et 40 ans (3% de formes juveniles et 16% tardives).
De nombreux patients ont des enfants et ont transmis leur gene a une autre generation soit par
transmission de gene soit par mutation spontanee.
II. Épidémiologique
Sex ratio: 1 (autant homme que femme). 1 sur 10000 affecté (0, 01%) soit environ 6000 cas en
france. 3 personnes sur 10000 sont porteuses du gene. 12000 personnes seraient porteuses de la
mutation mais asymptomatique. Prevalence moindre en chine, japon et finlande dû a un nombre
moindre des alleles genetiques avec répétition du CAG (cytosine, adenine, guanine). La MH est la
plus frequente des maladies rares.
=> Retombée sociales: moyenne de 5 personnes a risque soit concerne dans notre pays plus
de 100000 personnes (risque), cause de deces importante. La prise en charge est tres
importante dans cette pathologie quelle soit financiere, psychologique ou social.
3% mort par suicide
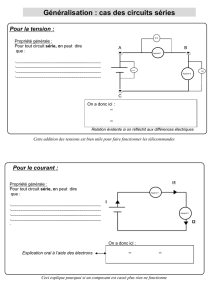
III. Genetique et rappel
Existence de 22 paires dautosomes (chromosomes non sexuels) et dune paire dheterosome
(heterochromosomes, chromosomes sexuels, gnosomes). Les chromosomes sont homogenes d'où
2 exemplaires de chaque gène, homologues mais pas toujours identiques. On parle dindividus
homozygotes (monozygotes) quand les 2 alleles sont identiques.
Caryotype: 22 autosomes et 2 heterosomes.
Quand 2 alleles, 2 situations:
Gene dominant: un seul s'exprime (secrit en majuscule).
Gene recessif (minuscule).
Gene condominant: les 2 s'expriment.
Plusieurs genotypes (gene) possibles pour un seul phénotype (yeux marrons). On donne le
numéro du chromosome au niveau du genotype.
La MH est une maladie genetique autosomale (pas chromosome sexuel) ie 50% des enfants
affectes.

1 ll 2 3 ll 4
1 ll 3 1 ll 4 2 ll 3 2 ll 4
1. Genetique
En 1983 localisation du chromosome 4. En 93, identification du gene responsable (4p16.3), cest
le gène code pour une protéine (huntingtine) compose dune sequence de 3 nucleotides CAG. Dans
le cas de la MH répétition anormale soit plus de 26 fois (moins de 26 aucune manifestation ni
risque, de 27 a 35 pas de manifestation mais risque de transmission, de 36 a 39 faible penetrance
et risque de transmission de moins de 50%, de 40 a 59 manifestation adulte et risque de 50%, plus
de 60 forme juvenile).
2. Diagnostic genetique et antenatal
Le diagnostic genetique necessite du consentement eclaire, dune equipe spécialisée (rapport
benefice risque, vivre avec ou sans diagnostic). Le diagnostic antenatal
(diag pre implantatoire, amniocenthese). Le test genetique est interdit pour les mineurs sauf sil y a
suspicion de declenchement de certains symptomes.
3. Neuropathologie
Degenerescence des noyaux gris centraux (planification des mouvements complexes et role dans
le demarrage et larret des mouvements). Atrophie bilaterale du Striatum, caracteristique la plus
precoce et la plus larque (60% datteinte du putamen et noyau caude) et atteinte du pallidum.
Forme juvénile jusque 90%.
Destruction neuronale avec 2 gradients devolution:
Postero anterieur (atteinte primitive des regions postérieures du putamen qui progressent
vers lavant en respectant partiellement le noyaux accumbens)
Oblique (débute dans les régions dorsales du noyaux caude et du putamen et qui setend
vers les régions ventrale et laterale).
Latrophie des ganglions de la base est présente chez les porteurs du gene bien avant le debut des
symptomes moteurs. Il faut attendre X % de neurones touches avant les symptomes.
=> On est en sous cortical, atteinte corticale? Precoce?

IV. Essais thérapeutiques
Neuroreconstruction: greffe intracerebrale de neurones foetaux donc transplantation
dans le striatum de cellules foetales, plusieurs essais.
Facteur neurotrophique et neuroprotection: protection des neurones contre les
phénomène de dégénérescence (ciliary neurotrophic factor CNTF).
1
/
4
100%