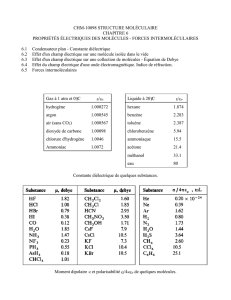
Chapitre 2 – Caractérisation des états de la matière

1
Chapitre 2 – Caractérisation des états de la matière – Christian Scheiber
PARTIE 1 : ETAT GAZEUX
I – Introduction
A) Thermodynamique Classique et Statistiques
- La Thermodynamique, c’est l’étude des systèmes macroscopiques en terme d’échange d’énergie
et/ou de matière avec le milieu extérieur.
- Il existe deux types de thermodynamiques :
- Classique : qui décrit les relations entre les différentes propriétés macroscopiques.
- Statistique : qui explique les lois de la mécanique à l’échelle microscopique.
- La Thermodynamique repose sur deux principes fondamentaux :
- La conservation de l’énergie.
- Le sens de la transformation.
B) Le Système Thermodynamique
- Voici les différents systèmes dans lesquels on étudie la Thermodynamique :
Système
Echange de matière
Echange d’énergie
Isolé
-
-
Fermé
-
+
Ouvert
+
+
- Lorsqu’il n’y a pas de transfert de chaleur, on parle de système adiabatique.
- Q = 0 : Adiabatique
- Q > 0 : Apport de Chaleur (Selon la convention du Banquier)
- Q < 0 : Perte de Chaleur (Selon la convention du Banquier)
- On établit les relations entre des Variables d’état qui peuvent être de différentes natures :
- Extensive (V, m, Concentration…)
Dépend de la quantité de matière
- Intensive (P, T)
Ne dépend pas de la quantité de matière.
- On ne s’intéresse qu’aux systèmes thermodynamiques chimiques.
C) Etat d’équilibre
- Etat de repos macroscopique qui est atteint spontanément par un système abandonné à lui-même.
- Les variables macroscopiques pour définir un état d’équilibre ont des valeurs fixes, définies et
mesurables.
D) Equation d’état
- Les différentes variables ne sont pas toutes indépendantes. En effet, il existe une relation entre le
Pression (« P »), le Volume (« V ») et la Température (« T ») : La Relation des Gaz Parfaits.
-
Avec n = Quantité de Matière (en Mole)
T = Température (en Kelvin)
R = Constante des gaz Parfait = 8,314J.mol-1.K-1
P = Pression (en Pascal)
V = Volume (en m3)

2
E) Transformation
- Réversibles : - Etat très proche de l’équilibre
- Les conditions extérieures doivent changer peu.
- Transformation lente.
- Irréversibles : - Se déroule spontanément dans la nature.
II – Température & Chaleur
A) Température et Chaleur
- La Température est une notion subjective qui découle du sens du toucher. Ce qu’on ressent ce
n’est pas une différence de Température, mais de Chaleur que l’on évacue.
B) Echelle de température
- La mesure de la température nécessite une échelle. Celle-ci est historiquement fait n fonction de la
température de Fusion et d’ébullition de l’eau à Patm. Tf = 0°C et Teb = 100°C.
C) Equilibre thermique
- Par conduction, le chaud se refroidit et vice versa jusqu’à égalité des températures.
D) Principe zéro de la thermodynamique.
- Si deux corps sont en équilibre thermique avec un troisième corps, alors ils sont en équilibre
thermique entre eux.
E) Chaleur & Equilibre thermique
- La chaleur, c’est l’énergie transférée par des chocs moléculaires désordonnés.
- L’équilibre thermique c’est lorsque le transfert de chaleur (d’énergie donc !) s’arrête.
F) Rapidité du transfert de chaleur.
- La rapidité du transfert de chaleur dépend de la paroi qui sépare un système de l’extérieur.
- Lorsque l’on est en présence d’une paroi adiabatique, il n’y a pas de transfert de chaleur.
- Lorsque l’on est en présence d’une paroi diathermique, il y a un transfert rapide de chaleur.
III – Gaz Parfaits et Réels
A) Modèle du gaz parfait
- Un gaz parfait est un état idéal vers lequel tendent les gaz à densité faible.
- Les molécules sont des points matériels qui n’ont pas d’énergie interne et qui ne développent par
conséquence qu’une énergie cinétique (énergie dépendant d’un mouvement.) Ces molécules vont
sous l’effet de leur mouvement venir se heurter à la paroi du système et créer une force que l’on
appelle la Pression.
- L’ensemble des variables d’état des gaz parfaits sont liées par la relation des gaz parfaits
développée précédemment :
B) Température absolue d’un gaz parfait
- L’énergie cinétique est la température. En effet, la température se définit à l’état microscopique (on
le verra plus loin) comme le degré d’agitation des molécules. Ce degré d’agitation augmente lorsque
l’on augmente l’apport en énergie. Or, l’énergie cinétique est la seule énergie exprimée par les
molécules !
- Lorsque l’on travaille sur les gaz parfaits, la pression est peu dense, peu changeante. Ainsi, il
est préférable de confronter les deux variables d’état : Température et Volume et de supposer la
Pression comme constante.

3
- Si on effectue un diagramme avec ses 2 variables, on peut extrapoler et prévoir la valeur de la
Température pour un volume nulle (= -273,15°C). Cette valeur correspond dès lors au zéro absolu est
vaut 1K !
C) Echelle de Température
- A partir du constat précédent, on peut lier les deux unités de la Température (K et °C) par la
relation :
- On rappelle que le Kelvin est utilisé comme unité du SI. Il doit, par conséquent, être utilisé pour la
relation des gaz parfaits et dans la constante des gaz parfait (8,314 J.K-1.mol-1)
- La relation PV = NRT génère plusieurs autres lois. En effet, on peut considérer dans certains cas que
la Pression, la Température ou le Volume soit constant (Pas les 3 à la fois !!!). On obtient dès lors :
- La loi de Boyle Mariotte (Isotherme) : PV = Constant.
- La loi de Gay-Lussac (Isobare) : V = V0 . (1 + αt) ou
- La loi de Charles (Isochore) :
= Constant
- Application : Volume occupée par une mole de gaz parfait à 20°C = 22,4 L
= 2,24.10-4m3
E) A l’échelle microscopique
- A cette échelle, nous n’utilisons plus :
- La constante des gaz parfaits mais la constante de Boltzmann.
- Le nombre de moles mais le nombre de molécules.
SelonThomsonàl’échellemicroscopiquelorsquel’onapporteuntravailApportdeChaleur
ledegréd’agitationdesmoléculess’élèveestc’estcequiexpliquel’augmentationdelatempérature
En effet, on rappelle que l’énergie cinétique = Température.
F) Equivalence microscopique et macroscopique
- On peut passer de la formule macroscopique (PV=nRT) à la formule microscopique (PV=NKT) en
utilisant le nombre d’Avogadro Na = 6,02.10-23 molécule.mol-1.
G) Température et énergie moléculaire.
- La température et l’énergie moléculaire sont des approches statistiques. En effet, on ne peut pas
prendre en compte les vitesses et l’énergie cinétique de chaque molécule d’un échantillon. On fait
alors, une approximation en cherchant l’énergie cinétique moyenne.
- La vitesse de chaque molécule n’étant pas, pour les mêmes raisons, calculable, on va partir du fait
que les molécules ont la même masse et ainsi déterminer (en prenant en compte l’énergie cinétique
moyenne) la vitesse quadratique moyenne.
-
-

4
Vqmoy = Vitesse quadratique moyenne. C’est une information physique sur ce qui se passe dans le
gaz. Comment se déplacent en moyenne les gaz d’un échantillon ?
H) Théorie cinétique des gaz parfaits. La Pression – Energie interne.
- Lorsque une molécule se translate dans un système, il est possible que celle-ci rencontre les
molécules de la paroi et l’exercice d’une force apparait.
- L’ensemble des forces exercées sur la paroi du système par les molécules est une force moyenne
que l’on nomme la pression
- Nous pouvons aller plus loin et lier la Pression au nombre de molécules, à leur masse, au volume
occupé et leur vitesse par la relation suivante :
- De plus, comme la molécule n’a pas d’E potentielle, ETOT (ou Ut) = EC =
- Enfin, on peut déterminer que
Or, PV = NkT
=
-
I) Energie cinétique moyenne et Vitesse quadratique moyenne.
- La constante de Boltzmann vaut la
=
= 1,38.10-23
-
-
J) Distribution des vitesses moléculaires de H2
- Dans un échantillon, les molécules ne présentent pas toutes un seul et unique état. En effet, elles
ne sont pas toutes identiques en terme de déplacement, d’énergie interne, de vitesse de translation
(ainsi que vibration et rotation pour les gaz réels).
- Cependant, il est raisonnable de penser que, dans une condition définie (Température), il y a un
état qui est prédominant. C’est pour cela que nous étudions la distribution de la loi de Maxwell
Boltzmann (voir partie K.)
- Sur la courbe suivante qui illustre la distribution de Boltzmann, on peut observer la quantité de
molécules pour une vitesse donnée. On cherche alors : A telle vitesse, combien a-t-on de molécules ?
A terme, on cherchera à connaitre la valeur de la vitesse pour laquelle on a un maximum de
molécules, dans des conditions de températures définies.
Dans le cas où T = 2000K, la vitesse moyenne des molécules se situe entre 3750 et 4500 m.s-1.

5
- On constate de plus que quand la température augmente :
- le nombre maximum de molécules diminue
- la courbe s’élargit.
K) Loi de Boltzmann
- La loi de Boltzmann permet de déterminer le nombre na de molécules qui ont une quantité
d’énergie Ea à une température T.
-
On constate que plus Ea est élevée, plus na est faible.
- Selon les molécules, le niveau d’énergie varie grandement.
L) Mélange de gaz parfait
- Lors d’une association de gaz parfaits :
- Les gaz n’interagissent pas.
- on peut calculer la pression partielle de chaque gaz telle que : PV = nRT
M) Loi de Dalton
- Selon la loi de Dalton, la Pression totale exercée sur les parois du système est égale à la somme des
pressions partielles des gaz constituant le mélange présent au sein du système.
- Autrement dit,
- De plus, dans le calcul de la relation des gaz parfaits, ntotal = ∑ ngaz
Application de la loi de Dalton :
- Quelle est la valeur de la PO2 au niveau de la mer et à une altitude de 7000m où P=0,45 atm ?
Données : Dans une mole d’air sec, on a 0,21 moles d’O2 Valeur constante jusqu’à 80 km d’alt.
Réponses
1) Au niveau de la mer : P = 1 atm = 2,1.104 Pa
2) A 7000m : P = 0,45 atm = 9450 Pa
N) Solubilité gaz – Loi de Henry
- La loi de Henry nous permet de relier la concentration d’un gaz dissous dans une solution et sa
pression partielle dans l’air expiré.
- Avec qui dépend : - Nature du gaz et liquide
- La température
Application de la loi d’Henry :
- Lorsqu’un plongeur descend trop profondément, il respire une concentration beaucoup trop
importante de Diazote ce qui provoque la maladie des caissons.
O) Les Gaz réels
- Dans les gaz parfaits, ils n’existaient pas certaines forces intermoléculaires qui sont pourtant
présentes chez les gaz réels. On peut dès lors élargir l’équation des gaz parfaits en prenant en
compte ces variations et en instaurant une nouvelle équation : l’équation de Van der Walls :
Avec : -
= Pression supplémentaire due aux interactions entre les molécules.
- b = Volume des molécules (Covolume)
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
1
/
21
100%