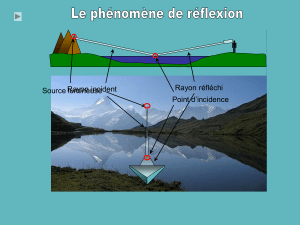
fluoroacrylate de 2,2,2 trichloroéthyle et de l

UNIVERSITÉ DU MAINE
LCOM-CHIMIE DES POLYMÈRES
U.F.R. Sciences et Techniques Unité de Chimie Organique Moléculaire et
Macromoléculaire, UMR CNRS n°6011
THÈSE
Pour obtenir le grade de
DOCTEUR
Spécialité : Chimie et Physico-Chimie des Polymères
Par
Jean-Marc CRACOWSKI
Synthèse et caractérisation de (co)polymères halogénés
pour l’optique linéaire -
Application à la réalisation d’un guide d’onde
Soutenue le 22 décembre 2008 devant le Jury composé de :
Mr Claude BUNEL Professeur, INSA-Rouen Rapporteur
Mr Thierry HAMAIDE Professeur, Université Lyon 1 Rapporteur
Mr Bruno AMÉDURI Directeur de Recherche, ENSC de Montpellier Examinateur
Mr Dominique BOSC Ingénieur de Recherche, Université de Rennes 1 Examinateur
Mr Régis MERCIER Chargé de Recherche, Université de Savoie Examinateur
Melle Véronique MONTEMBAULT Maître de Conférences, Université du Maine Encadrante
Mr Fabrice ODOBEL Directeur de Recherche, Université de Nantes Directeur de thèse
Mr Laurent FONTAINE Professeur, Université du Maine Directeur de thèse

« Le génie commence les beaux ouvrages, mais le travail seul les achève ».
Joseph Joubert

Le travail présenté dans ce mémoire a été réalisé dans l’Unité de Chimie Organique
Moléculaire et Macromoléculaire de l’Université du Maine (UMR CNRS n° 6011) grâce au
soutien financier de la Région Pays de la Loire et le soutien du CNRS.
Je tiens à exprimer ma profonde reconnaissance à Messieurs Claude Bunel,
Professeur à l’INSA de Rouen, et Thierry Hamaide, Professeur à l’Université de Lyon 1, qui
m’ont fait l’honneur d’accepter de juger ce travail en tant que rapporteurs. J’associe à ces
remerciements Messieurs Régis Mercier, Chargé de Recherche au CNRS à l’Université de
Savoie, Dominique Bosc, Ingénieur de Recherche à l’Université de Rennes 1, et Bruno
Améduri, Directeur de Recherche au CNRS à l’Ecole Nationale Supérieure de Chimie de
Montpellier, pour leurs précieux conseils et pour avoir accepté de participer à ce jury.
Je remercie vivement mon directeur de thèse, le Professeur Laurent Fontaine, pour
l’accueil qu’il m’a réservé au sein de son équipe, pour la confiance qu’il m’a accordé, pour
ses conseils avisés, pour les discussions scientifiques que nous avons eu ainsi que pour
m’avoir donné un aperçu du rôle et du travail (ingrat) d’un directeur de laboratoire.
J’exprime toute ma reconnaissance et mon amitié sincère à Mademoiselle Véronique
Montembault pour m’avoir encadré durant ces trois années, et particulièrement pour sa
gentillesse, sa disponibilité, la rigueur qu’elle m’a enseignée ainsi que pour ses nombreuses
corrections au stylo rouge qui me traumatiseront à vie. J’exprime également ma gratitude à
Monsieur Fabrice Odobel, Directeur de recherche à l’Université de Nantes, pour avoir
accepté de co-encadrer cette thèse.
Je tiens à remercier mon tuteur pédagogique, le Docteur Albert Laguerre, pour
m’avoir encadré lors de mon monitorat. Grâce à ses conseils, j’ai pu pleinement apprécier la
fonction d’enseignant à l’université et j’ai pris beaucoup de plaisir à effectuer cette tache.
Je remercie grandement ma maman au labo, Anita Loiseau, ainsi que Fred Legros et
Jean-Luc Moneger pour leur aide au quotidien, et également Martine et Cécile pour les
nombreuses (oh oui très nombreuses) analyses RMN.

Mes plus sincères salutations vont à l’ensemble des permanents du laboratoire qui
m’ont beaucoup apporté tant professionnellement que personnellement: Jean-François (dit
papa), Fred, Irène, Charles I, Jean-Claude, Daniel (allez le MUC), Sagra et Michel.
Mes remerciements vont également à mes collègues doctorants du laboratoire, qui ont
su m’accueillir lors de mon arrivée et avec qui j’ai fait quelques soirées mémorables. Grâce à
eux j’ai pu garder le moral au beau fixe : Chuanpit, Anuwat, Nitinart (et Alisa), Ekkazit,
Ekkawit, Fon, Tang moo, Doan, Hoa, Gnoc, Dien, Thomas, Sandie, Charles (dit Carlito),
Faten, Gaëlle et Rachid. Merci à mes amis thaïlandais et vietnamiens avec qui j’ai pu
découvrir d’autres cultures (et d’autres nourritures), je sors particulièrement enrichi au
niveau personnel de ces rencontres. Merci particulièrement à mon ami Rachid, heureusement
que tu étais là, cette thèse n’aurait pas eu la même saveur sans toi (une saveur de Kristal
weissen !). Tous vous allez me manquer, j’ai vraiment apprécié ces trois ans passés en votre
compagnie.
Evidemment ces remerciements ne pourraient être complet sans une pensée pour mes
deux grands-mères, j’espère que de la haut vous êtes fières de moi. Merci à ma famille d’être
là et d’avoir fait de moi ce que je suis. Je vous aime. Et un gros bisou à mon Bichou pour
m’avoir supporté pendant ces trois ans.
Enfin fervent supporter de l’OL, je remercie cette équipe pour le 8
ème
titre qu’elle va
bientôt nous offrir, et surtout je remercie vivement l’Olympique de Marseille qui nous fait
bien rire.

 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
1
/
198
100%