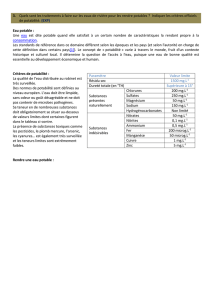
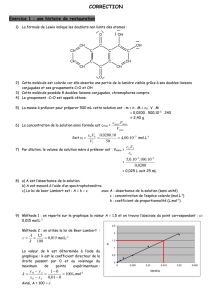
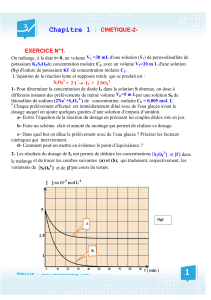
Propriétés inhibitrices d`un mélange d`amines grasses et de

______________________________________________
THÈSE
Présentée pour obtenir le grade de
Docteur de l’Institut National Polytechnique de Toulouse
Ecole Doctorale : Sciences de la matière
Spécialité : Science et Génie des Matériaux
par
SRISUWAN Nakarin
Propriétés inhibitrices d'un mélange d'amines grasses et de sébaçate
de sodium vis-à-vis de la corrosion d'un acier au carbone
Soutenue publiquement le 27 Juin 2008
Composition du jury :
Mme SUTTER Eliane Professeur à l’IUFM de varsailles, Université de Cergy-Pontoise Rapporteur
M. NORMAND Bernard Professeur à l’ INSA de Lyon Rapporteur
M. MORAN Francis CEFRACOR, Paris Examinateur
M. LAMURE Alain Professeur à l’I.N.P de Toulouse Examinateur
M. TRIBOLLET Bernard Directeur de recherche au CNRS, Paris Examinateur
Melle PEBERE Nadine Directrice de recherche au CNRS, Toulouse Directrice de thèse
____________________________________________________________________________________________
Institut Carnot CIRIMAT – UMR CNRS 5085
ENSIACET, 118 route de Narbonne, 31077 Toulouse Cedex 04
i

Résumé
Ce travail porte sur l’inhibition de la corrosion d’un acier au carbone dans une solution de NaCl
200 mg.L-1 par une formulation inhibitrice non toxique utilisée pour le traitement des eaux des
circuits de refroidissement. Elle est constituée d’amines grasses (AG) et de sébaçate de sodium
(SS). Le tracé des courbes de polarisation et la spectroscopie d’impédance électrochimique ont
été utilisées afin de caractériser le mode d’action de chaque inhibiteur et d’optimiser les
concentrations de chacun des composés dans le mélange. Un maximum d’efficacité a été
obtenu en présence de 50 mg.L-1 AG + 1,5 g.L-1 SS. Un mécanisme d’adsorption coopérative
des deux composés explique l’effet de synergie observé. Le film inhibiteur est constitué d’un
mélange d’oxyde/hydroxyde de fer incorporant les molécules organiques. La détermination des
énergies de surface a confirmé la mécanisme d’adsorption coopérative proposé à partir des
mesures électrochimiques.
Abstract
This work is devoted to the corrosion inhibition of a carbon steel in a 200 mg.L-1 NaCl solution
by an original multi-component inhibitor used for water treatment in cooling circuits. The
inhibitive formulation was composed of fatty amines (FA) in association with sodium sebacate
(SS). Steady-state current-voltage curves were combined with electrochemical impedance
measurements to characterise the inhibitive properties of each compound and to optimize the
concentration of the compounds in the mixture. Maximum efficiency was reached for the
mixture containing 50 mg L-1 FA + 1.5 g L-1 SS. A cooperative adsorption mechanism was
proposed to account for the observed synergistic effect. The inhibitive film was composed of an
iron oxide/hydroxide mixture incorporating the organic molecules. Surface energies
determination comborate the cooperative adsorption mechanism shown from the
electrochemical tests.
ii

A mes parents en témoignage de tout mon amour, à toute ma famille et à
ma très chère grand-mère pour m’avoir accompagné dans cette grande aventure.
i

ii

Remerciements
En préambule à ce manuscrit, je souhaite exprimer ma reconnaissance et mes remerciements
aux personnes qui m’ont soutenu et aidé durant les années consacrées à la réalisation de ce
travail au Centre Interuniversitaire de Recherche et d’Ingénierie des Matériaux à l’ENSIACET.
Dans un premier temps, j’exprime ma gratitude au ministère des affaires étrangères, du
gouvernement français et au King Monkut’s University of Technology North Bangkok, Thai-
French Innovation Centre en Thaïlande pour avoir soutenu financièrement mon travail de thèse.
Je tiens à remercier Monsieur Bernard Tribollet, Directeur de recherche au CNRS au
Laboratoire interfaces et systèmes électrochimiques (LISE) à Paris pour toutes les
connaissances qu’il m’a transmises dans le domaine de l’électrochimie et aussi pour avoir
accepter de présider mon jury de thèse.
Je tiens à remercier sincèrement Madame Eliane Sutter, Professeur à l’IUFM de varsailles,
Université de Cergy-Pontoise et Monsieur Bernard Normand, Professeur à l’ INSA de Lyon
pour avoir accepté d’être rapporteurs de ce travail. Leurs commentaires et suggestions ainsi que
les différents échanges que nous avons eu m’ont été d’une grande aide dans la finalisation de ce
travail.
J’adresse un remerciement particulier à Monsieur Alain Lamure, Professeur à l’I.N.P de
Toulouse pour sa participation au jury de thèse comme examinateur. Il m’a beaucoup appris
dans le domaine des mesures de l’énergie de surface.
J’exprime ma gratitude à monsieur Francis Moran, ancien Président Directeur Général de la
société de Concorde Chimie, pour s’être déplacé afin de participer à mon jury de thèse en tant
qu’examinateur. J’ai été très heureux de sa présence.
Mes plus grands remerciements vont à Mademoiselle Nadine Pébère, Directrice de recherche
au CNRS, qui a été ma directrice de thèse. Je ne saurai jamais assez la remercier pour l’énergie
qu’elle a dépensé afin que je puisse réaliser ce travail. Je la remercie aussi pour l’autonomie
iii
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
1
/
123
100%