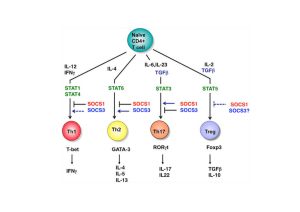
PDF - Université Paul Sabatier

T
TH
HÈ
ÈS
SE
E
En vue de l'obtention du
D
DO
OC
CT
TO
OR
RA
AT
T
D
DE
E
L
L’
’U
UN
NI
IV
VE
ER
RS
SI
IT
TÉ
É
D
DE
E
T
TO
OU
UL
LO
OU
US
SE
E
Délivré par l'Université Toulouse III - Paul Sabatier
Discipline ou spécialité : Immunologie
JURY
Pr Roland LIBLAU Président
Dr Maria Cristina CUTURI Rapporteur
Pr Jean DAVOUST Rapporteur
Pr Joost van MEERWIJK Directeur de Thèse
Ecole doctorale : Biologie - Santé - Biotechnologies
Unité de recherche : U563 Inserm
Directeur(s) de Thèse : Joost van Meerwijk
Présentée et soutenue par
Thibault SANTOLARIA
Le 26 janvier 2009
Titre : INDUCTION DE TOLERANCE AUX ALLOGREFFES D'ORGANES SOLIDES PAR LES
LYMPHOCYTES T REGULATEURS CD4+ CD25+ FOXP3+

1

2
REMERCIEMENTS
Aujourd’hui se tourne une page importante de ma vie, quatre années passées
à l’Inserm U563 et pourtant, quand je regarde en arrière, j’ai l’impression d’être
seulement arrivé hier. Cette page est émminamment importante puisque c’est elle
qui m’a vu, pour rester dans l’immunologique, me différencier de cellule immature
(étudiante) en cellule effectrice, apte à la dure loi de la sélection du travail… Même si
une telle transformation demande une intégration personnelle du message (« La
chimère hématopoïétique est à la réponse à toutes les questions » et autre « Les
Tregs vont sauver le monde »), elle ne saurait s’opérer sans une éducation et un
signal de survie de la part d’autres cellules spécialisées (la grande famille des Chefs,
et la famille tout court).
Dans mon cas, ma cellule présentatrice d’antigènes est certainement mon
directeur de thèse : Joost van Meerwijk. Merci à vous de m’avoir accepté au sein du
DEA d’immunologie, puisque c’est ainsi que cela s’appelait encore, et de m’avoir
associé à l’un des plus passionnants sujets qui soit : la tolérance aux autres. Merci
également de m’avoir tout au long de ces quatre années enseigné l’art délicat de la
chimère hématopoïétique, la passion des Tregs et le goût de former la génération
suivante. Je garderai en mémoire, de nos « interactions », les longues semaines,
pleines de doutes et de craintes, à patienter pour savoir si oui ou non, nos greffes de
coeur allaient survivre jusqu’à terme ; les mois à attendre que nos coupes
histologiques soient enfin prêtes ; les jours précédents l’envoi du papier à faire et
refaire, encore et encore, des photos avec Talal ; et votre détermination sans faille
pour défendre le papier devant l’Editorial Board de Nature Medicine. Peu y aurait cru,
vous oui ! Encore moins aurait réussit un tel pari, vous si ! Le jeu en valait la
chandelle… Merci encore pour ses intences émotions.
Si dans cette métaphore immunologique, Joost devait être la CPA, alors Paola
serait certainement la mTEC. Parfait complément dans cette étape de sélection, vous
avez su m’apporter la rigueur nécessaire à la réalisation d’un projet scientifique.
Merci pour votre disponibilité, les nombreux rapports de congrès que vous nous avez
fait (et les discussions qui s’ensuivaient) et votre enthousiasme au quotidien. Mon
seul regret sera que vous ne m’ayez que partiellement communiqué votre passion
pour la sélection thymique. Il faut dire que je partais avec un a priori assez fort vis-à-
vis de cette partie de l’immunologie.
Merci à Geneviève pour sa gentillesse et son aide précieuse dans le
phénotypage des nombreuses souris transgéniques utilisées au laboratoire.
Bien sûr, le thymocyte n’est pas seul dans l’immensité du thymus. De très
nombreux autres lymphocytes en devenir l’accompagnent dans cette longue
transformation. Sans eux, le pauvre thymocyte ne serait plus le même. Parmi eux, il y
a d’abord mes collègues de l’équipe Tolérance et Auto-immunité, à commencer par

3
REMERCIEMENTS
les anciens, qui par leur sagesse ont contribué à ma formation. Un grand merci donc
à toi, Olive, pour m’avoir appris les bases techniques du sujet et donné tous les
conseils dont j’avais besoin : de l’étude comparative entre les pansements Carrefour
et Hansaplast pour les greffes de peau, à la technique secrète de l’ACK ou les
chemins détournés pour traverser Toulouse en pleine heure de pointe pour rentrer
chez mes parents. Merci enfin, pour ta patience, devant mon style parfois si
particulièrement particulier (« Lors du dysfonctionnement d’un organe, celui-ci
n’assure plus sa fonction »). Ingrid, pendant les trois ans que je t’ai cottoyée, tu as
été un tel soleil, de par ta fougue, ta gentillesse, et ton caractère si trempé, qu’il est
difficile de t’oublier. Tantôt souriante, tantôt attendrissante quand tu parlais de Tristan
et parfois si effrayante quand tu t’enflammais contre quelqu’un, tes rires resonneront
encore longtemps contre les murs du labo. Mille mercis pour ce que tu es ! Surtout
ne change pas… Julie, tu es la seule à m’avoir accompagné tout au long de ces
quatre ans. D’abord intimidé par tes « Allo » si engageant, j’ai rapidement découvert
derrière le masque, une fille extrêmement attachante et toujours prête à aider. Nos
discussions, tant sur le Stade Toulousain que sur les derniers exploits d’un Ministre
appelé à devenir Président à la place du Président, me manqueront. Merci
également pour tes idées débordantes pour fêter le départ d’un des notres… idées
qui m’ont tour à tour, mis dans la peau de Mitch, Travolta ou Charlie et ses drôles de
dames. Enfin, comment oublier la petite Julie Ribot, mascotte de l’équipe et
ambassadrice des pauses cafés-clopes. Même dans les moments difficiles, tu m’as
toujours soutenu, écouté et je t’en remercie.
Place maintenant aux jeunes, à la relève du labo. Honneur à la première
arrivée : Céline ! C’est maintenant toi la doyenne du labo, le pillier sur lequel
compteront les futurs étudiants. Merci pour ton écoute, tes discussions souvent
enflammées et ton entrain quotidien. A Lise, je t’ai refilé le bébé et espère qu’il t’e
donnera autant de bonheur et de réussite qu’il m’en a apporté. Bon courage pour la
suite et profite bien de ces quelques années de thèse : elles ont une saveur toute
particulière. Clémence, nos chemins ne se sont malheureusement pas croisés très
longtemps mais cela m’a suffit pour comprendre qu’un peu à l’image d’Ingrid, tu allais
rapidement devenir la coqueluche de l’U563, grâce à ta sympathie et à ta bonne
humeur quotidienne. Mais, j’ai également découvert que, comme elle, tu n’étais pas
genre à te faire marcher sur les pieds. Entre toi, Lise et Céline, Joost a donc du souci
à se faire. Peut-être en fait, devrais-je lui souhaiter à lui « Bon Courage ». Enfin, il est
temps de boucler la boucle, celle-là même qui avait commencé quand Olive m’avait
cédé sa place comme seul coq de la basse-cour et qui se termine aujourd’hui
puisque je laisse Svet au milieu de ces filles. Cela peut faire peur de prime abord,
mais tu verras : on y prend goût très vite. En tout cas, courage pour la suite de ton
M2R !

4
REMERCIEMENTS
Je remercie également tous les membres de l’unité, les anciens comme les
nouveaux, pour avoir partagé avec moi tant de bons moments pendant ces 4
longues années : Florie, Camille, Sabina, Loïc, Nico, Yovan, Delphine, Lucile, Audrey
et Patrick de l’animalerie, Fatima, Dominique, notre super secrétaire, et Aline, notre
deuxième mère. Une mention spéciale à Nicolas, mon talentueux partenaire de
dance, à Sophie, la gentillesse incarnée, à Aurelie, la « people » de l’U563, à Julie
Tabiasco, la pile électrique toujours prête à aider, et Michaël, l’animateur de nos
repas de midi, qui, chacun dans son style, m’a beaucoup apporté.
Pour terminer, je remercie mes parents sans qui, je ne serais pas celui que je
suis aujourd’hui et qui m’ont toujours soutenu ; mes amis qui depuis déjà 13 ans pour
certains, me supportent et me permettent d’oublier les petits tracas de la vie. Enfin, si
je devais une dernière fois utilisée la comparaison avec l’immunologie, je dirais
qu’une cellule effectrice ne saurait exister sans sa contrepartie régulatrice, chacune
étant le versant opposé de l’autre, unies en un tout... N’est-ce pas la définition de
l’âme sœur ? Merci à toi Karine d’être ma cellule régulatrice, celle qui fait mon
bonheur et mes peines et qui, comme moi, a subit la parfois difficile sélection
thymique.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
1
/
168
100%