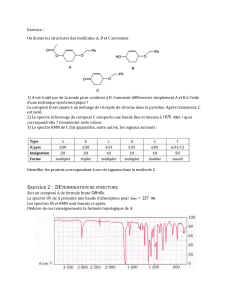
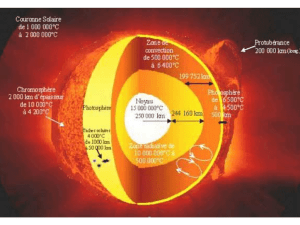
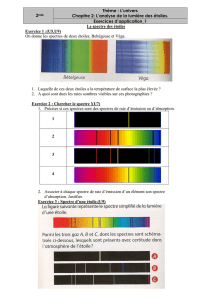
polycopié Chim 12 - Université Kasdi Merbah Ouargla

Méthodes Spectroscopique (Chim.12). 3
MINISTERE DE L’ENSEIGNEMENT SUPERIEUR ET DE LA RECHERCHE
Université KASDI MERBAH Ouargla
Faculté des Mathématiques et des Sciences de la Matière
3
ième
Année Licence
S
PECTROSCOPIQUES
Méthodes Spectroscopique (Chim.12). 3
ième
année Licence chimie analytique (UKMO) K.DEHAK
MINISTERE DE L’ENSEIGNEMENT SUPERIEUR ET DE LA RECHERCHE
SCIENTIFIQUE
Université KASDI MERBAH Ouargla
Faculté des Mathématiques et des Sciences de la Matière
Département de Chimie
DEHAK Karima
Année Licence
: Chimie Analytique et Chimie Organique
METHODES
PECTROSCOPIQUES
(Chim.12)
1
année Licence chimie analytique (UKMO) K.DEHAK
MINISTERE DE L’ENSEIGNEMENT SUPERIEUR ET DE LA RECHERCHE
Faculté des Mathématiques et des Sciences de la Matière
: Chimie Analytique et Chimie Organique
PECTROSCOPIQUES

2
Méthodes Spectroscopique (Chim.12). 3
ième
année Licence chimie analytique (UKMO) K.DEHAK
Sommaire
GENERALITES SUR LE SPECTRE ELECTROMAGNETIQUE…………….…........................... 4
CHAPITRE I : SPECTROPHOTOMETRIE UV-VISIBLE………………………...………………
4
I.1.Introduction …………………………………………………………..........................................
4
I.2.Principe…………………………………………………………………………
.
.……………… 4
I.3.Spectre d’absorption UV-Vis……………………………………………………........................ 5
I.4.Principaux types de transitions électroniques ……………………..…………………………... 5
I.5.Analyse quantitative……………………………………………………..……………………… 6
I.6.Analyse qualitative…………………………………………………………..……...................... 7
CHAPITRE II: SPECTROMETRIE DU PROCHE ET DU MOYEN INFRAROUGE…….............
11
II.1.Introduction …………………………………………………………………............................. 11
II.2.Origine de l’absorption lumineuse dans l’IR………………………….…………………….….
11
II.3.Spectre d’absorption dans l’IR.......………………………………………….............................. 11
II.4.Modes de vibration ……………….……………………………………………......................... 11
II.5.Application de l’I.R à la
détermination des diverses fonctions d’un composé
organique…………………………………………………………………………………………….
12
II.6.Exemples de spectres IR de composés organiques…………………………………………….. 13
II.7.Appareillage……………………………...…………………………………………………….. 15
CHAPITRE III : SPECTROSCOPIE DE RESONANCE MAGNETIQUE NUCLEAIRE
(RMN)..................................................................................................................................................
17
III.1.Introduction…………………………………………………………………………………….
17
III.2.Types d'échantillons…………………………………………………………............................ 17
III.3.Principe………………………………………………………………………............................ 17
III.4.Transitions entre ces niveaux d’énergie……………………………………………………….. 18
III.5.Appareillage………………………………………………………………….............................
19
III.6.Résultat d'une analyse RMN…………………………………………………........................... 19
III.7.Le déplacement chimique……………………………………………………............................ 20
III.7.Blindage des noyaux ………………………………………………………………………….. 21
III.8.Intégration des signaux………………………………………………………………………… 22
III.9.Facteurs affectant les déplacements chimiques ………………………………………………. 23
III.10.Phénomène de couplage spin-spin……………………………………………………………. 24

3
Méthodes Spectroscopique (Chim.12). 3
ième
année Licence chimie analytique (UKMO) K.DEHAK
CHAPITRE IV : SPECTROMETRIE DE MASSE (SM)……………………………….................. 26
IV.1.Introduction……………………………………………………………………………………
26
IV.2.Principe ……………………………………………………………………………………….. 26
IV.3.Spectre de masse ……………………………………………………………............................ 27
IV.4.Principaux procédés d’ionisation……………………………………………............................
27
IV.5.Exemple d’application en spectrométrie de masse……...……………………………………...
28
IV.6.Fragmentation de quelques molécules organiques…………………………………………….. 29
CHAPITRE V: SPECTROMETRIE D’ABSORPTION ATOMIQUE ET EMISSION DE
FLAMME ……………………………………..........................................................................…….
34
V.1.Introduction………………………………………………………………….............................. 34
V.2.Origine de l’absorption et de l’émission……………………………………............................... 34
V.3.Principe………………………………………………………………………............................. 35
V.4.Dosage par absorption ou par émission de flamme…………….………………………………. 37
V.5.Appareillage……………………………………………………….…………............................. 37
V.7.Utilisation pratique dans le cas le plus classique………………………..…………………….... 39
REFERENCES ……………………………………………………………………………………..
40

Méthodes Spectroscopique (Chim.12). 3
Généralités sur le spectre électromagnétique
Le spectre électromagnétique est l’ensemble continu des ondes électromagnétiques connues et
classées soit par leurs
fréquence
leurs énergies
∆E. Il est constitu
Figure 1
: Principales régions du spectre électromagnétique
La spectrométrie d’absorption est basée sur les interactions entre la matière et le rayonnement
électromagnétique.
Ce rayonnement est constitué de photo
Deux cas sont possibles, ce photon peut réagir soit
valence) ou avec les électrons
les molécules se traduit par l’absorption de quanta d’énergie, ce qui conduit au passage de ces
molécules de leur
état énergétique fondamental à un état excité
plus élevée : E = h
Chaque rayonnement conduit à un type d
correspond une méthode spectroscopique.
Exemples
Rayons X
: électrons des couches internes
UV-Vis
: excitation des électrons de valence
IR : déformation des liaisons
Radio fréquence
: RMN (spin nucléaire)
CHAPITRE I-
SPECTROPHOTOMETRIE UV
I.1.Introduction
La spectroscopie UV-
Vis est la plus ancienne et la plus utilisée des méthodes d’analyse. Elle
permet notamment des applications quantitatives. Cependant, elle ne fournit que peu
d’informations structurales (Analyse qualitative) comparées aux autres méthode
spectroscopiques (IR, RMN et SM).
I.2.Principe
L’absorption d’un photon dans le domaine UV
électronique
de la molécule, alors que les termes des énergies
sont moins affectées.
Méthodes Spectroscopique (Chim.12). 3
ième
année Licence chimie analytique (UKMO) K.DEHAK
Généralités sur le spectre électromagnétique
Le spectre électromagnétique est l’ensemble continu des ondes électromagnétiques connues et
fréquence
s ν, leurs longueurs d’ondes , leurs
nombre
E. Il est constitu
é par l
es grandes régions électromagnétique
: Principales régions du spectre électromagnétique
La spectrométrie d’absorption est basée sur les interactions entre la matière et le rayonnement
Ce rayonnement est constitué de photo
ns porteurs de quanta d’énergie.
Deux cas sont possibles, ce photon peut réagir soit
avec
les électrons de liaisons (électrons de
valence) ou avec les électrons
des couches internes d’un atome.
L’effet du rayonnement
les molécules se traduit par l’absorption de quanta d’énergie, ce qui conduit au passage de ces
état énergétique fondamental à un état excité
,
lequel possède une énergie
Chaque rayonnement conduit à un type d
’excitation propre à lui et à chaque excitation
correspond une méthode spectroscopique.
: électrons des couches internes
: excitation des électrons de valence
: RMN (spin nucléaire)
SPECTROPHOTOMETRIE UV
-
VISIBLE
Vis est la plus ancienne et la plus utilisée des méthodes d’analyse. Elle
permet notamment des applications quantitatives. Cependant, elle ne fournit que peu
d’informations structurales (Analyse qualitative) comparées aux autres méthode
spectroscopiques (IR, RMN et SM).
L’absorption d’un photon dans le domaine UV
-
Vis provoque une augmentation de
de la molécule, alors que les termes des énergies
rotationnelle et vibrationnelle
4
année Licence chimie analytique (UKMO) K.DEHAK
Généralités sur le spectre électromagnétique
Le spectre électromagnétique est l’ensemble continu des ondes électromagnétiques connues et
nombre
s d’ondes σ ou
es grandes régions électromagnétique
s suivantes :
: Principales régions du spectre électromagnétique
La spectrométrie d’absorption est basée sur les interactions entre la matière et le rayonnement
ns porteurs de quanta d’énergie.
les électrons de liaisons (électrons de
L’effet du rayonnement
sur
les molécules se traduit par l’absorption de quanta d’énergie, ce qui conduit au passage de ces
lequel possède une énergie
’excitation propre à lui et à chaque excitation
VISIBLE
Vis est la plus ancienne et la plus utilisée des méthodes d’analyse. Elle
permet notamment des applications quantitatives. Cependant, elle ne fournit que peu
d’informations structurales (Analyse qualitative) comparées aux autres méthode
s
Vis provoque une augmentation de
l’énergie
rotationnelle et vibrationnelle

Méthodes Spectroscopique (Chim.12). 3
Notons que :
E
tot
= E
L’absorption
de photons se traduit par des transitions d’électrons engagés dans les orbitales
moléculaires situées à la frontière entre les derniers niveaux occupés
liaisons σ et π ai
nsi que les non liants n)
occupés des états excités (σ
*
longueur d’onde λ
max
et par son coefficient d’absorption molaire
d’onde.
I.3. Spectre d’absorption UV
IL s’agit du diagramme représentant l’absorption d’une radiation UV
l’échantillon à étudier en fonction de la longueur d’onde de la radiation.
Les deux paramètres les plus importan
d’onde des maximas d’absorption (
d’absorption molaires Є
max
.
Figure I.1 : S
pectre UV
I.4.Principaux types de transitions électroniques
Les transitions les plus rencontrées
n π* et n σ*.
-
Transition *
La grande stabilité
des liaisons
énergétique important entre les niveaux
effet, cette transition est intense située dans le lointain UV, vers 130 nm.
Exemple : éthane : λ
max,
Les hydrocarbures saturés tels que l’hexane ou le cyclohexane qui ne présentent que
des liaisons σ sont pratiquement transparents dans le proche UV et peuvent de ce fait
être utilisés comme solvants pour des composés organiques peu polaires ou apolaires.
- Transition
π π*
Les composés possédant une double liaison éthylénique isolée conduisent à une forte
bande d’absorption vers 170 nm. (Exemple
Abs.
Méthodes Spectroscopique (Chim.12). 3
ième
année Licence chimie analytique (UKMO) K.DEHAK
E
élec
+
E
rot
+E
vib
de photons se traduit par des transitions d’électrons engagés dans les orbitales
moléculaires situées à la frontière entre les derniers niveaux occupés
nsi que les non liants n)
de l’état fondamental et les premiers niveaux non
*
et π
*). Chaque transition est caractérisée à la fois par sa
et par son coefficient d’absorption molaire
: Є
max
I.3. Spectre d’absorption UV
-Vis
IL s’agit du diagramme représentant l’absorption d’une radiation UV
l’échantillon à étudier en fonction de la longueur d’onde de la radiation.
Les deux paramètres les plus importan
ts sur un spectre UV-Vis sont les
valeurs des longueurs
d’onde des maximas d’absorption (λ
max
) et des épaulements (λ
ép
) ainsi que les coefficients
pectre UV
-Vis du -
carotène dans le chloroforme
max1
= 466nm et
max2
= 497nm)
I.4.Principaux types de transitions électroniques
Les transitions les plus rencontrées
dans les composés organiques sont:
σ
*
:
des liaisons
σ des compos
és organiques se traduit par un écart
énergétique important entre les niveaux
σ (O.M liante) et σ* (O.M antiliante). En
effet, cette transition est intense située dans le lointain UV, vers 130 nm.
max,
= 135 nm, Є
max
= 10 000.
Les hydrocarbures saturés tels que l’hexane ou le cyclohexane qui ne présentent que
σ sont pratiquement transparents dans le proche UV et peuvent de ce fait
être utilisés comme solvants pour des composés organiques peu polaires ou apolaires.
π π*
:
Les composés possédant une double liaison éthylénique isolée conduisent à une forte
bande d’absorption vers 170 nm. (Exemple
: éthylène λ
max,
= 165 nm,
λ
max
1
λ
λ
max
2
5
année Licence chimie analytique (UKMO) K.DEHAK
de photons se traduit par des transitions d’électrons engagés dans les orbitales
moléculaires situées à la frontière entre les derniers niveaux occupés
(les électrons des
de l’état fondamental et les premiers niveaux non
*). Chaque transition est caractérisée à la fois par sa
max
à cette longueur
IL s’agit du diagramme représentant l’absorption d’une radiation UV
-Vis traversant
valeurs des longueurs
) ainsi que les coefficients
carotène dans le chloroforme
σ
σ*, π π*,
és organiques se traduit par un écart
σ* (O.M antiliante). En
effet, cette transition est intense située dans le lointain UV, vers 130 nm.
Les hydrocarbures saturés tels que l’hexane ou le cyclohexane qui ne présentent que
sont pratiquement transparents dans le proche UV et peuvent de ce fait
être utilisés comme solvants pour des composés organiques peu polaires ou apolaires.
Les composés possédant une double liaison éthylénique isolée conduisent à une forte
= 165 nm, Є
max
= 16 000).
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
1
/
40
100%