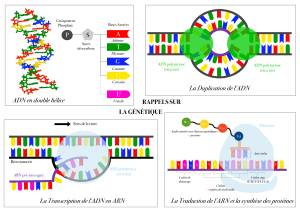
Maintient et remodelage de l`information génétique. Comment les

Maintient et remodelage de l’information génétique.
Comment les organismes vivants perpétuent leurs potentiels génétiques ? Pour affronter les
attaques sur leurs génomes ? Pour générer de la diversité ?
Importance de check points sur les mécanismes 3R de l’ADN.
Qu’est ce qu’une ADN polymérase ?
Enzyme qui rajoute des trinucléotides en utilisant une matrice (exception pour la terminale
polymérase qui n’en utilise pas). Copie dans le sens 5’-3’. Pour initier le phénomène, il faut une
amorce en bout de chaine d’ADN.
Si la polymérase rajoute de nombreux trinucléotides, on l’appelle une polymérase processive,
dans le cas contraire, il s’agit d’une polymérase peu processive. Une polymérase qui respectera
correctement les complémentarités A-T et C-G sera une polymérase fidèle. Mais des fois, la
polymérase peut mettre un C en face d’un A etc. Il existe des tests de fidélité. Mais beaucoup de
ces enzymes sont également capable de défaire ce qu’elles ont fait : il s’agit d’une relecture avec
une activité exonucléasique de 3’-5’.
Lorsque les polymérases arrivent face a des dimères de thymines (induits sous rayonnement
UV), elles s’arrêtent, incapables de reconnaitre ces dimères. Mais certaines peuvent baille-passer
le problème : ce sont des polymérases translésionelles (elles sont peu fidèles).
Une ADN polymérase est un complexe enzymatique intervenant dans la réplication de l’ADN
au cours du cycle cellulaire, mais aussi dans des processus de réparation et de recombinaison de
l'ADN. Il existe différentes familles de polymérases qui diffèrent selon leurs séquences en acide
aminé et leurs propriétés catalytiques.
Toutes les ADN polymérases synthétisent l’ADN dans le sens 5'-3' et aucune n’est capable de
commencer une nouvelle chaîne sans amorces. Elles ne peuvent que rajouter des nucléotides à
partir d’une amorce préexistante à l’extrémité 3’-OH. Pour cette raison l’ADN polymérase a
besoin d’une amorce (ou primer), sur laquelle elle pourra ajouter de nouveaux oligonucléotides.
L’amorce peut être formée d’ADN ou d’ARN et est synthétisée par une autre enzyme, appelée
primase. Une enzyme, hélicase, est ensuite requise pour délier le double brin de l’ADN et ainsi
faciliter l’accès des ADN polymérases sur les brins d’ADN, devenu simple brin, et permettre
ainsi la réplication. Les ADN polymérases possèdent une structure très conservée. Elles sont
considérées comme étant des holoenzymes puisqu’elles ont besoin d’un ion magnésium comme
cofacteur pour fonctionner correctement. En absence d’ions magnésium, elles sont appelées
apoenzymes.
Les ADN polymérase ont la capacité de corriger les erreurs dans la formation de brin néoformé.
Lorsqu'une paire de base incorrecte est reconnue, l’ADN polymérase va revenir en arrière grâce
à son activité 3’-5’ exonucléase, va réinsérer la base correcte, et reprendre la réplication.
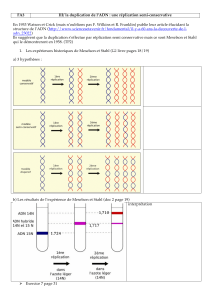
Expériences de mutagénèse chez E. coli.
Le mutant Pol I a une activité polymérase amoindri mais la bactérie vit très bien => paradoxe.
On a ainsi découvert les différents types de polymérases chez la bactérie.
Cinq ADN polymérases ont été identifiées chez les bactéries:
Pol I : impliquée dans la réparation de l’ADN. Elle possède les deux activités
polymérase 5'→3' et exonucléase 3'→5' (fragment de Klenow), et participe à la synthèse
des fragments d’Okazaki. Elle intervient aussi en fin de réplication pour éliminer les
amorces d'ARN (activité exonucléase 5'-3').
Pol II : impliquée dans la réplication de l'ADN endommagée et possède activité 5'→3' et
une activité a 3'→5' exonucléase.

Pol III : c’est la principale polymérase bactérienne qui intervient dans l'élongation de la
chaîne d'ADN lors de la réplication au niveau du brin avancé et de la synthèse des
fragments d’Okazaki. Elle est constituée de dix sous-unités. On définit une structure
minimale (core enzyme αεθ) comprenant une sous-unité α (activité polymérase), une
sous-unité ε (exonucléase 3'→ 5') et une sous-unité θ de fonction inconnue. Deux cores
(αεθ) et un complexe γ (facteur de chargement) sont maintenus ensemble par
l'intermédiaire d'un connecteur, la protéine t.
Pol IV : ADN polymérase de la famille Y.
Pol V : ADN polymérase de la famille Y.
Pour chacun des brins, il existe une synthèse continue pour l’un, et une discontinue pour l’autre
(avec utilisation des fragments d’Okazaki).
Chez un mutant Pol.1-, comment peut-on obtenir des mutants spécifiques de la réplication ?
On va utiliser des mutants conditionnels (thermosensibles…).
On a ainsi observé 2 types de mutants :
-les mutants Quick Stop (QS) : arrêt immédiat de la réplication de l’ADN avec une
modification de la température.
-les mutants Slow Stop (SS) : arrêt lent de la réplication de l’ADN avec une modification
de la température.
Chez les QS : identification d’un certain nombre de gènes intervenant dans la réplication tels que
Pol III (avec plusieurs sous unités), Pol II… ; et pour les SS, on a identifié des gènes codant pour
des protéines impliquées dans le cycle de réplication.
La polymérase I (Pol I).
Activité polymérasique dans le sens 5’-3’, exonucléasique dans le sens 5’-3’ (la seule qui fait ca)
et donc naturellement aussi exonucléasique dans le sens 3’-5’. Peu processive. % d’erreur très
faible.
La polymérase III (Pol III).
Activité polymérasique dans le sens 5’-3’, exonucléasique dans le sens 3’-5’. Processivité
importante, très dépendante de la sous unité ξ. % d’erreur très faible.
La vitesse de réplication chez E. coli est d’environ 1000 nucléotides/seconde. Le génome d’E.
coli comporte 4Mbases.
Les cœurs de la polymérase sont essentiels au phénomène de polymérisation : sous unités α, ξ, θ.
Si ξ est inactive, le % d’erreur est multiplié par 1000, mais la vitesse reste inchangée.
La primase est importante pour les fragments d’Okazaki. Tandis que l’hélicase sépare les deux
brins d’ADN. La polymérase agit sous forme d’homodimère, chaque dimère étant associée à l’un
des brins. La sous unité β, structure en anneau, homodimère, fait gagner en processivité. Le trou
laissé dans l’anneau du dimère permet le passage du brin d’ADN.
Fragment d’Okasaki est un terme utilisé en biologie, dans l'étude des chromosomes.
Le brin « retardé » est synthétisé par petits fragments (d’environ 10 paires de bases) : les
fragments d’Okasaki.
L’ADN polymérase pour effectuer la synthèse d’ADN nécessite une amorce (petit fragment
d’ADN ou d’ARN). Cette amorce ARN est amenée par l'enzyme ARN Primase qui est une

enzyme ARN polymérase ADN dépendante.
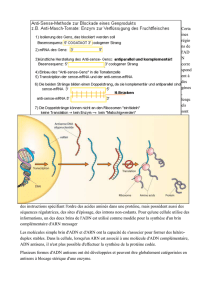
Description: Depiction of DNA replication with replication fork, strands and okazaki-fragments.
a: template strands, b: leading strand, c: lagging strand, d: replication fork, e: primer, f: Okazaki
fragment
Entre les deux, il existe un gros complexe important : le clamp-loader qui pose l’anneau β au bon
endroit (clamp-loader = complexe θ-γ). Codage : le même gène donne deux produits protéiques
différents : τ et τ1. C’est un régulateur de la progression de la fourche. Ce complexe τ est
important pour la fourche de réplication en agissant avec l’hélicase, qui est poussée en amont de
la fourche de réplication.
Le clamp-loader va déplacer une des deux sous unités β qui va, sur le brin simple laissé (car une
des deux polymérases ne peut pas synthétiser de façon continue), synthétiser le fragment
d’Okazaki, puis qui sautera de quelques milliers de bases pour de nouveau synthétiser un
fragment d’Okazaki etc.
La primase est une ARN polymérase qui fabrique de petites amorces de ribonucléotides. La Pol I
prend le relais de la Pol III pour combler le trou entre les fragments d’Okazaki. Pol I prends la
suite de la polymérisation de Pol III, puis avec son activité exonucléasique détruit l’ARN pour
faire de l’ADN. Il faut ensuite rétablir la continuité des brins avec la ligase.
La primase est une enzyme permettant la synthèse de l'amorce d'ARN nécessaire à la synthèse
du brin d'ADN au cours de la réplication de l'ADN. Les ADN polymérases permettant la
réplication de l'ADN ont besoin en effet d'une petite région double-brin pour commencer la
synthèse d'ADN, et cela dans les trois domaines du vivant (Archaea, eucaryotes, Bacteria).
La primase des bactéries est DnaG. Chez les eucaryotes, elle est portée par le complexe ADN
Polymérase α-primase. Chez les Archaea, il existe un homologue à la primase eucaryote, mais
sans équivalent à la sous-unité polymérase α. Il est surprenant de noter que les primases
d'Archaea peuvent également synthétiser de l'ADN in vitro. Il existe également chez les
Archaea un homologue à la protéine DnaG des bactéries.
Initiation de la réplication chez E. coli.

Il existe des séquences de 13 ou 9 paires de bases à l’origine de l’oriC (origine de réplication).
Rentrée en jeu d’une hélicase fabriquant une petite amorce. Phénomène de méthylation pour
synthétiser les brins et éviter une autre réplication successive (la méthylation prend environ 20-
30min, déterminant ainsi le temps de réplication de l’ADN et donc la division bactérienne).
Les séquences Ter recrutent des protéines tus. Lorsque l’hélicase arrive dans ces séquences Ter,
la protéine tus est absente, et cela permet la transcription. Fixation spécifique de tus. Ces
protéines tus permettent le passage de la Pol qui arrive dans un sens, mais pas dans l’autre (utile
dans le cas ou la fourche de réplication n’arrive pas à la même vitesse [car la réplication du
chromosome bactérien est bidirectionnelle, et la fin de la réplication est variable sur le
chromosome, en fonction de la vitesse des polymérases de chacun des deux cotés]) [La protéine
tus agit comme une véritable diode moléculaire, permettant le passage des Pol dans un sens, mais
pas dans l’autre].
Chez les eucaryotes :
On retrouve des équivalents des mêmes structures (PCNA = équivalent des anneaux β)
[homotrimère de taille identique que l’anneau β qui lui est un dimère]
La polymérase est une ARN-ADN polymérase dépendante. Synthèse de quelques nucléotides et
le reste du complexe prends en charge de l’ADN. Les fragments d’Okazaki sont plus courts que
chez les bactéries. Il existe des centaines d’origine de réplication.
Pour diminuer le petit fragment d’ARN entre les fragments d’Okazaki, on a les RNAses I qui
coupent le petit bout de l’amorce et FEN1 qui dégrade le bout d’ADN par activité
endonucléasique.
Maintient et remodelage de l’information génétique.
Plus l’organisme est simple, moins son ADN comporte de paires de bases. Chez les amphibiens
et les insectes, il y a une grande variabilité de la quantité d’ADN. Certains insectes ont jusqu'à
100x plus d’ADN que d’autres insectes.
E.coli : 4Mbases Drosophile : 100Mbases
Alors, quelle est l’information contenue dans l’ADN ? Notion de complexité de l’ADN, et non
plus de taille. Certains ADN seront informatifs, d’autres peu informatifs.
Pour déterminer cela, on dénature puis on le fragmente. On va ensuite regarder les propriétés de
renaturation. Un brin peu informatif aura beaucoup de répétitions de nucléotides. Par exemple,
l’ADN ayant un poly-A aura un énorme potentiel de réassociation, contrairement a de l’ADN
informatif => Il y a différente cinétique de renaturation. (Mesurable par absorption de la lumière
UV par les bases : simple brin et double brin n’ont pas la même absorbance.)
C : concentration de l’ADN simple brin au temps t
K : constante de renaturation.
Renaturation : dC/dt= -kC²
On intègre entre t et t0 => C /C 0 = 1 / (1+ k C 0 t)
=>...
=> C 0 t1/2 = 1/t
Résultats :

L’ADN de E. coli est beaucoup plus informatif que celui des bactériophages M32 et T4.
Chez les eucaryotes, on trouve des degrés de complexité variable dans le génome, avec des
séquences hautement répétées, peu répétées, ou unique. Les gènes se retrouvent majoritairement
dans la zone d’ADN non répétée.
Séquençages des génomes par la méthode de Sanger.
Il faut séquencer x fois les génomes pour éviter les erreurs de séquençage.
Génome de S. cerevisae.
-eucaryote : peu d’intron (3-4% des gènes)
-taille : 12Mbases sur 16 chromosomes.
-codant à 72%
~6200 ORF (CDS) [peu de gènes à introns] [environ 300]
Désormais, on a des techniques de séquençages plus performants
Ex : méthode du shotgun
Séparation des chromosomes -> clonage de grands fragments (YACs, BACs…) -> sous clonage
-> séquençage -> assemblage (1)
Clonage shotgun (plasmides) -> séquençage -> assemblage (2)
Chez l’homme, on utilise des marqueurs CA pour faire le séquençage car les motifs CA sont
répartis uniformément dans tout le génome. On peut faire des relations entre la carte
phénotypique et la carte génétique d’un chromosome. La recombinaison est d’autant plus active
dans les télomères que dans les centromères. La recombinaison est plus active chez la femme
que chez l’homme. Plus le bras du chromosome est long, moins la recombinaison est active
ADN satellite :
-ADN satellite localisé : ADN de faible complexité mais répété de nombreuse fois. Peut
représenter plusieurs % du génome (homme : 5-6% ; rat : >50% ; bœuf : 20%)
-ADN satellite dispersé : trouvé à peu prés n’ importe où dans le génome. On distingue
les microsatellites (<6 bases) des minisatellites (>6 bases)
 6
7
8
9
10
11
12
13
14
15
16
6
7
8
9
10
11
12
13
14
15
16
1
/
16
100%