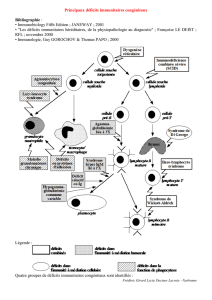
Les déficits immunitaires héréditaires

Les déficits immunitaires héréditaires
1- Généralités.
A) Distinction entre déficit héréditaire et déficit acquis.
Le déficit immunitaire héréditaire ou primitif peut être lié à une mutation de gènes impliqués dans le
développement et le fonctionnement des cellules immunitaires ou à une infection chez l’enfant jeune (sauf déficit
immunitaire commun variable ou DICV). Il induit donc une anomalie de la réponse immune, ainsi un diagnostic
précoce permet un traitement approprié dans certains cas. En l’absence de traitement on observe une aggravation
du déficit.
Le déficit immunitaire acquis ou secondaire est la conséquence d’autre pathologie (cancer), de facteurs
environnementaux, d’anomalie métabolique ou d’infection (VIH).
B) Classement des déficits immunitaires héréditaires.
Les déficits immunitaires héréditaires sont des maladies génétiques rares dont la prévalence est de 1/5 000
naissance (150 DIH sont décrits avec pour la majorité un gène identifié). Les déficits immunitaires héréditaires sont
classés en fonction du défaut immunologique biologique :
- Les déficits immunitaires combinés touche à la fois l’immunité cellulaire (LT) et humorale (LB) et +/- les NK.
- Les déficits de l’immunité humorale isolée.
Ces derniers sont tous deux des déficits héréditaires de l’immunité adaptative.
- Les déficits du complément.
- Les déficits des cellules phagocytaires (polynucléaires, monocytes et macrophages).
- Autres déficits de l’immunité innée.
Ces trois derniers sont des déficits héréditaires de l’immunité innée.
Les conséquences sont une prédisposition majeure aux infections.
C) Quand évoquer un déficit immunitaire héréditaire ?
Un déficit immunitaire héréditaire doit être évoqué devant :
- Des infections récurrentes des voies respiratoires hautes et basses : > 8 otites/an chez les moins de 4 ans, > 4
otites/an chez les plus de 4 ans, > 2 pneumonies/an ou > 2 sinusites/an.
- Des infections sévères à pneumocoque, Haemophilus, Neisseria : un seul épisode de méningite ou sepsis se
doit d’être exploré.
- Des infections à bactéries pyogènes récurrentes (cutanée, invasive, tissulaire).
- Des infections inhabituelles et/ou d’évolution inhabituelle (infection par un germe opportuniste, diarrhée
infectieuse persistante, muguet ou candidose cutanée récidivante).
- Une cassure de la courbe staturo-pondérale et/ou une diarrhée persistante.
- Des antécédents familiaux de déficits immunitaires ou de signes cliniques similaires.
D) Explorations.
On peut être orienté vers un déficit immunologique si lors d’une infection bactérienne on retrouve un déficit
d’anticorps, de complément ou de phagocytes, ou si lors d’une infection virale ou fongique on retrouve un déficit en
lymphocytes T.
Les examens de 1ère intention consistent en une NFS, une électrophorèse des protéines sériques, un dosage des Ig
et une sérologie post-vaccinale.
Les examens complémentaires de 2ème intention correspondent à un phénotype lymphocytaire, l’exploration des
fonctions lymphocytaires et phagocytaires, l’exploration du complément et la réalisation de test génétiques et
moléculaires.

2- Les déficits immunitaires combinés sévères (DICS) ou déficit complet de l’immunité adaptative.
A) Anomalie de la recombinaison : phénotype T- B- NK+.
Elle correspond à une anomalie de recombinaison des gènes des immunoglobulines et du TCR, c’est un déficit
immunitaire combiné sévère se caractérisant par un défaut de différenciation des lymphocytes T et B et donc par
une agammaglobulinémie type alymphocytose. On distingue deux types de déficits :
- Déficit spécifique de la lignée lymphoïde : Rag1/Rag2.
- Déficit affectant également d’autres tissus (expression ubiquitaire, voie NHEJ) : artemis, cernunos et ligase IV
avec une sensibilité cellulaire excessive aux radiations ionisantes.
La transmission dans ce cas est autosomique récessive, l’incidence est de l’ordre de 1/500 000 naissances.
NFS
Autres données
Diagnostic
Lymphopénie profonde, diminution des LT et LB
(voire absence)
NK normaux
HypoIgG, IgA et IgM
Atrophie précoce
thymique
Absence des différentes populations
lymphocytaires
Les conséquences sont la survenue d’infections opportunistes sévères et d’infections récurrentes bactériennes ou
virales (pneumocystose, candidose, CMV) vers le 3ème mois, de diarrhée et d’un retard staturo-pondéral.
Dans tous les cas face à un tel déficit il faut prohiber l’utilisation de vaccins vivants ainsi que la transfusion de
produits sanguins non irradiés.

Le traitement passe par la greffe de cellules de moelle osseuse ou de sang de cordon (létal en l’absence), et par le
développement de protocole de thérapie génique.
B) Défaut de signalisation cytokinique : phénotype T- B+ NK-.
Type d’interleukine
Effets et actions
IL-2
Régulation de la prolifération des LT activés
Tolérance T périphérique par induction de l’apoptose
Développement, survie et maintenance des Treg
Prolifération des LB
Production des Ig
IL-4
Régulation de la prolifération des LT activés
Différenciation des Th2
Production des Ig et commutation
IL-7
Développement des cellules T
Homéostasie des cellules T naïves
Survie des cellules T mémoires
Régulation de la recombinaison V(D)J
IL-9
Régulation de la prolifération des LT activés
Production des Ig
IL-15
Régulation de la prolifération des LT activés
Inhibition de l’apoptose T périphérique
Développement, survie et maintenance des LTCD8+
Développement et survie des NK
Expansion et homéostasie des NK
Activation des NK
IL-21
Favorise la formation des centres germinatifs par induction des LTCD4+ folliculaires
Différenciation et expansion clonale des Th17
Stimule les fonctions cytotoxiques des LTCD8+, des NK et des NKT
Prolifération et différenciation des LB
Induction de l’apoptose des LB en l’absence de signalisation par le BCR et les LT
Régulation de la production d’Ig et la commutation isotypique
Inhibition de la maturation et des fonctions des cellules dendritiques
a) Déficit de la chaine commune gamma (Rγc) des récepteurs aux cytokines.
C’est la forme la plus fréquente de déficit immunitaire combiné sévère, elle représente environ 45 à 50% des cas.
C’est un déficit récessif lié à l’X touchant 1/200 000 naissances.
NFS
Autres données
Diagnostic
Diminution des LT et NK
(voire absence)
LB normaux ou diminués
(parfois augmentés)
HypoIgG, IgA et IgM
Pas de développement thymique
Altération des fonctions des LB (par
absence d’activité T helper)
Absence d’expression de la chaine γc à la
surface des cellules hématopoïétiques
Les conséquences sont la survenue d’infections sévères et récurrentes dès les premiers mois de vie chez le garçon,
de diarrhée et d’un retard staturo-pondéral.
Le traitement passe par la greffe de cellules souches (seul traitement actuel de ce déficit immunitaire sévère).
b) Déficit en JAK3 (Janus Kinase 3).
La liaison des cytokines à leur récepteur entraine dans la situation normale une
autophosphorylation de JAK3 qui phosphoryle des résidus tyrosines sur son récepteur, d’où un
recrutement des STATs (facteurs de transcription) et une modulation de l’expression de gènes
cibles.

Le déficit en JAK3 représente 10% des déficits immunitaires combinés sévères, c’est une forme autosomique
récessive de DICS (chromosome 19).
Les signes cliniques classiques sont des manifestations au cours des premiers mois de la vie de diarrhée chronique ou
de retard de croissance.
Les manifestations cliniques, les données biologiques et le traitement sont identiques à ceux d’un déficit en γc.
c) Déficit en chaine α du récepteur à l’IL-7 (IL7-Rα).
Le récepteur à l’IL-7 (IL-7R) est formé d’un hétérodimère IL-7Rα (CD127) et d’une chaine γ commune (CD132). Lors
de la différenciation T (thymus), la signalisation médiée par l’IL-7 participe au réarrangement des gènes codant pour
le TCR (via une déméthylation de l’ADN et une acétylation des histones).
Dans toutes les cellules T, la signalisation via IL-7R conduit à l’activation des voies PI3K/AKT et STAT5 ce qui entraine :
- Une augmentation de l’expression de BCL-2 (anti-apoptotique) et une diminution de l’expression des
facteurs pro-apoptotiques BAX, BIM et BAD.
- Une diminution de l’expression de p27 (inhibiteur du cycle cellulaire) et une augmentation de l’expression de
CDC25A (activateur du cycle cellulaire).
Tout cela aboutit à une augmentation de la survie et de la prolifération des cellules T, de plus on observe une
prolifération TCR-indépendante pour les émigrants thymiques récents.
Le déficit en IL-7R est retrouvé dans moins de 5% des cas de déficit immunitaire combiné sévère, il est autosomique
récessif (chromosome 5). L’incidence est de 1/500 000 à 1/1 000 000 naissances.
NFS
Autres données
Diagnostic
Diminution des LT (voire absence)
LB et NK normaux ou diminués
LB non fonctionnels (absence de LT)
HypoIgG, IgA et IgM
Atrophie thymique
Absence d’expression d’IL7-Rα sur les blastes
(expression IL-2R, IL-4R, diagnostic différentiel du γc)
C) Déficits en adénosine déaminase (ADA) : phénotype T- B- NK-.
Un déficit en adénosine déaminase aboutit à une accumulation de métabolites toxiques entrainant la mort des
progéniteurs lymphoïdes.
C’est la seconde forme la plus fréquente de déficit immunitaire combiné sévère puisqu’elle représente 15% des cas
(1 à 2 naissance/an en France). L’incidence est de 1/200 000 naissances. C’est un déficit autosomique récessif.

NFS
Autres données
Diagnostic
Diminution des LT, LB et NK (voire
absence) donc lymphopénie
profonde
HypoIgG, IgA et IgM
Pas de développement thymique
(absence d’ombre thymique
radiologiquement)
Mesure de l’activité de l’ADA
érythrocytaire (en absence de
transfusion)
Les conséquences sont la survenue d’infections sévères et récurrentes (bactériennes, virales, fongiques) dès les
premiers mois de vie.
Le traitement est basé sur plusieurs thérapeutiques :
- Traitement substitutif par administration de l’enzyme couplée au polyéthylène-glycol.
- Allogreffe de cellules souches.
- Plusieurs tentatives de thérapie génique avec récemment une correction durable du déficit pour quatre
patients ayant bénéficié d’une greffe de leurs cellules souches génétiquement modifiées associée à une
myéloablation de faible intensité.
D) Autres déficits immunitaires combinés sévères : phénotype T- B+ NK+.
CD3 : γδε2ξ2
La signalisation par le TCR repose sur la transduction via CD3.
La phosphorylation des ITAM permet le recrutement des SRC
kinase (ZAP70, LCK, LYN et FYN).
CD45 : intervient dans
l’activation du LT par le
TCR
On retrouve CD45RA sur les LT naïfs et CD45R0 sur les LT
mémoires.
L’activité phosphatase cytoplasmique de CD45 enlève un
groupement phosphate inhibiteur de LCK, LYN et FYN libérant
leur activité tyrosine kinase.
Les déficits en CD3 et CD45 sont à l’origine de déficits immunitaires combinés sévères moins fréquents (uniquement
quelques cas décrits pour chacun des déficits). C’est un déficit autosomique récessif.
NFS
Autres données
Diminution des LT (voire absence), présence de LT immatures
(fonction de la chaine CD3 affectée)
LB et NK normaux
LB non fonctionnels (perturbation du signal helper)
HypoIgG, IgA et IgM
E) Résumé.
La fréquence et la transmission de ce déficit est de l’ordre de 1/100 000 (transmission autosomique récessive ou
récessive liée à l’X), sa révélation est précoce vers le 3ème mois de vie. En termes cliniques on retrouve des infections
opportunistes (pneumocystose), fongiques (candidose), virales (parainfluenzae, adénovirus, CMV), bactériennes ou
encore une BCGite.
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%