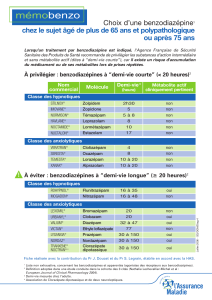
Notes de cours Pharmacologie : Biodisponibilité & Demi-vie
Telechargé par
Steve boris Ndole

Travaux dirigés Pharmacologie générale A3 ISVK
1. RAPPELS ET DEFINITIONS
1.1. Biodisponibilité du médicament
La biodisponibilité quantité
fraction de la dose
et à la vitesse !
"
"
!
" disponible
#Cmax”
$
#Tmax”
1.2. Demi-vie
% & ' & ( (
médicament' La demi)vie
médicaments
*+,-+,./0%"+1234./0%"
'&')
4&411')
51&4) '65&2,) '46'5
&34) '7'7&,8) '

Travaux dirigés Pharmacologie générale A3 ISVK
,
") &
$ 9
' : ;
)
1.3. Volume de distribution
/ le volume fictif # <
= >
%
?@$:
AU BILAN'!
• )neutres et liposolubles large et
homogène
• )'!
• )extracellulaire
• )intracellulaire
• $ ' (
!'résidus
(leurs délais d’attente

Travaux dirigés Pharmacologie générale A3 ISVK
4
2. APPRENTISSAGES
2.1. Exercice I
A&B 510C'
")
:
D1:4'85D:,'55D,:'25D8:1'65D7:1'5
Question N°1 : D
Question N°2 : ?D42
Question N°3 : %
Question N°4 ? E)
2.2. Exercice II
' 1 A 5 F'
51GH'
?&AF
Question N°1 : %
Question N°2 : %)
Question N°3 ? EAF

Travaux dirigés Pharmacologie générale A3 ISVK
8
2.3. Exercice III
@ ,5 51 C 1'5 0C 511
I0C 81 0C
J"):
D:7'55D,:8'25D8:'65D7:1'5
Question N°1 : %%
1
"?)
2.4. Exercice IV
"K' :
Absorption : A&' 31L"
! K
,'1'5
M &1,A
,11 B' % ! K 5'42
0"%1',70""AM%
! ,N7'82J0""
5151 514 51
Distribution : " 841 2"0C
61 C " K
L?
'
31L)!
AAH M AAH
Métabolisme :K'4L
!
8514A8
Elimination :") !3
Question N°1 :
OP >

Travaux dirigés Pharmacologie générale A3 ISVK
5
3. SOLUTION DES EXERCICES
Solution I
%")
A%AM%
%D4:0 %2:1',50
/?:0%
1
+5104'8+8'50C
D
0,
+'7%
A%AM%:4'850,+'6,0
 6
6
1
/
6
100%